


Controlo cinético e controlo termodinâmico
📧
- Universidade do Porto
Referência Corrêa, C., (2016) Controlo cinético e controlo termodinâmico, Rev. Ciência Elem., V4(2):016
DOI http://doi.org/10.24927/rce2016.016
Palavras-chave Controlo; cinética; termodinâmica; molécula; reação; fusão; ebulição;
Resumo
As reações químicas podem ser mais ou menos completas e mais ou menos rápidas. A Termodinâmica trata das questões de equilíbrio e a Cinética trata das questões de velocidade. Em muitos casos as reações mais completas são mais rápidas, mas muitas vezes as reações incompletas são rápidas e as completas são lentas.
O que é que realmente controla uma reação química?
1. Controlo cinético
Quando uma
reação é controlada cineticamente, os produtos que se formam mais
rapidamente obtêm-se em maiores quantidades. Por exemplo, na nitração do
tolueno, a 0ºC, obtêm-se os isómeros orto-, meta- e para- nitrotoluenos
em percentagens que refletem as respetivas velocidades de formação.
A nitração do tolueno é uma substituição eletrófila semelhante à nitração do benzeno. O agente de nitração é vulgarmente o catião nitroílo, NO2+, obtido da reação entre o ácido sulfúrico e o ácido nítrico (Fig. 1).
O ácido nítrico protonado perde uma molécula de água e origina o catião NO2+. A este nível elementar pode considerar-se que a substituição se efetua em dois processos elementares, o primeiro mais lento (limitante da velocidade de nitração) e o segundo, perda de um protão, muito rápido.
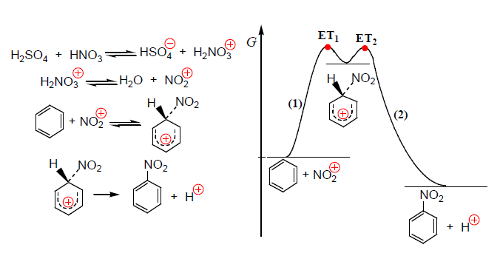
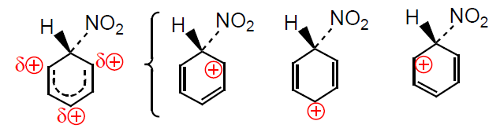
No primeiro processo forma-se um carbocatião estabilizado por deslocalização eletrónica, o intermediário de Wheland, com a carga positiva deslocalizada sobre o anel (Fig. 2). No processo (2) o protão sai por ação de HNO3 e HSO4−.
A nitração do tolueno é mais rápida do que a nitração do benzeno (o grupo CH3 é ativante e orto/para diretor), isto é, favorece a entrada do grupo nitro nas posições orto e para. A figura 3 mostra um diagrama correspondente à substituição.
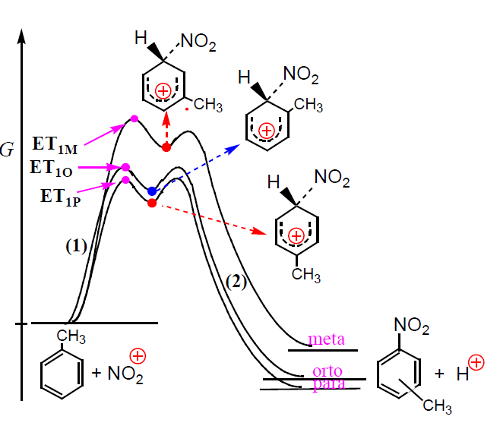
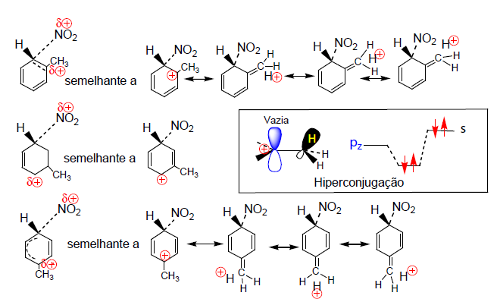
Os estados de transição dos processos (1), ET1M, ET1O e ET1P, conducentes aos intermediários nitrometilbenzénio são estabilizados do mesmo modo que os intermediários a que conduzem (Fig. 4). A estabilização processa por hiperconjugação, que consiste na interação da orbital pz do anel (vazia) com orbitais σ do grupo metilo, mas só ocorre quando o ataque se dá nas posições orto e para.
A velocidade de nitração do tolueno é cerca de 20 a 25 vezes maior do que a nitração do benzeno devido à presença do grupo metilo. A 0ºC, a distribuição de isómeros nitrotoluenos é a seguinte: o-nitrotolueno – 43%, m-nitrotolueno – 4% e p-nitrotolueno – 53%, o que correlaciona bem com as velocidades relativas de ataque nas mesmas posições (Fig. 5).
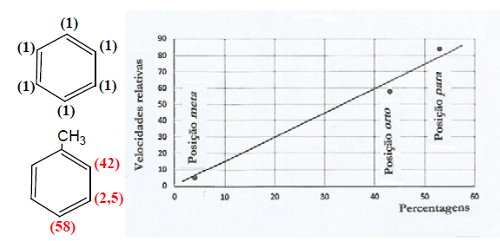
Esta reação de nitração é controlada cineticamente dado que as percentagens de isómeros são determinadas pelas velocidades relativas de reação.
2. Controlo termodinâmico
A
constante de equilíbrio de uma reação, K , está relacionada com a
variação da energia livre de Gibbs, ΔGº , pela expressão
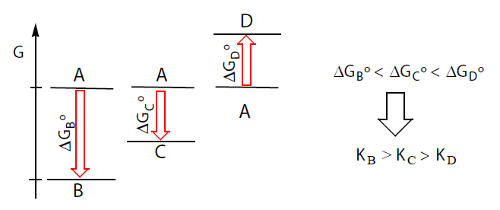
ΔGº = – RT ln K. Quanto menor (mais negativo) for ΔGº
mais completa é a reação. Assim, a reação A→B é mais completa que
a reação A→C que é mais completa que a reação A→D.
De um modo geral, para uma série de reações semelhantes, as variações de entropia, ΔSº, não diferem muito de reação para reação e, por isso, as variações de ΔGº dependem fundamentalmente das variações de ΔHº (ΔGº = ΔHº - TΔSº).
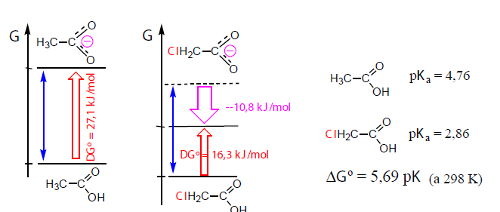
A extensão de muitas reações pode comparar-se com base na análise dos efeitos de estabilização/instabilização resultantes da introdução de átomos e grupos na molécula. Por exemplo, a protólise de ácidos carboxílicos é afetada pela presença de átomos e grupos dadores e atraidores de eletrões (Fig. 7).
Embora os dois ácidos tenham energias livres de formação diferentes, a saída de um protão do grupo carboxílico dos dois ácidos em meio aquoso deveria envolver uma igual variação da energia livre (a azul), quer dizer a energia livre do anião cloroacetato devia situar-se no nível tracejado da figura. No entanto, dada a capacidade do cloro para atrair eletrões, ou seja dado o seu efeito indutor negativo, a carga negativa do anião, em vez de se deslocalizar somente sobre o grupo carboxílico, é também dispersa sobre o cloro. Tal como a deslocalização de carga, a sua dispersão também contribui para estabilizar os sistemas. No caso da figura 2, a estabilização é de cerca de 10 kJ/mol. O átomo de cloro também exerce o seu efeito no estado inicial (ácido) mas o seu efeito é especialmente importante no anião, por ter uma carga elétrica.
Um exemplo mais elucidativo, por partir do mesmo composto inicial, é a protólise do ácido málico (ácido hidroxibutanodióico), que origina em maior percentagem o anião resultante da protólise do grupo carboxílico vizinho do grupo hidroxilo, por ter uma extra-estabilização ΔΔG por ação do grupo OH (fortemente atraidor de eletrões por efeito indutor negativo), o que sucede em menor grau quando o grupo OH está mais afastado (Fig. 8).
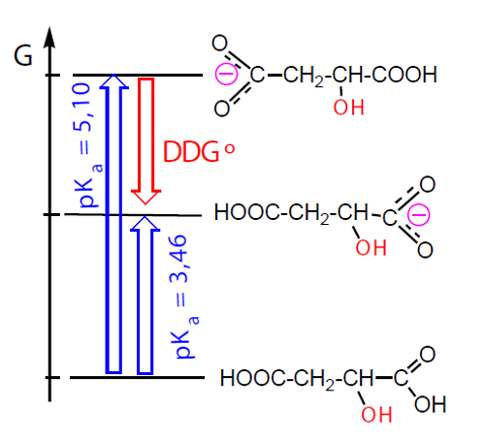
Neste caso, a relação entre as constantes de equilíbrio é a mesma que a relação entre as concentrações dos produtos formados o que significa que o controlo da reação é termodinâmico.
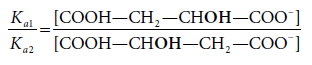
As reações de protólise de ácidos carboxílicos são bastante rápidas, e os produtos mais estáveis obtém-se em maiores quantidades. São reações controladas termodinâmicamente.
3. Controlo cinético versus controlo
termodinâmico
Nos exemplos apresentados encontrámos reações
controladas cineticamente e reações controladas termodinâmicamente.
Vamos agora analisar casos em que, para a mesma reação e para o mesmo
composto, o controlo cinético passa a termodinâmico e vice-versa,
conforme as condições da reação.
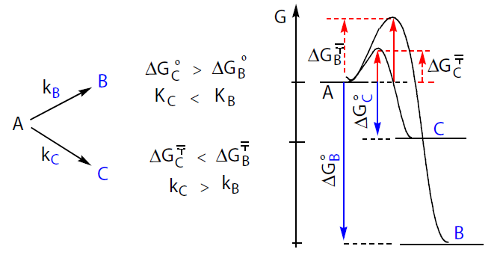
Consideremos uma reação em que uma substância A se pode transformar em B ou C segundo duas reações paralelas A → B e A → C. Pode suceder que a reação A → C seja mais rápida mas menos completa e que a reação A → B seja mais lenta mas mais completa.
Qual dos produtos, B ou C, se formará em maior quantidade? O controlo será cinético ou termodinâmico? A figura 9 resume a situação e apresenta um diagrama de energias para as duas reações.
Um caso típico desta situação é a adição de HBr a dienos conjugados em que a adição pode ocorrer nos carbonos 1 e 2 ou nos carbonos 1 e 4. A reação efetua-se via carbocatiões resultantes da adição rápida de H+ à ligação dupla terminal conforme se mostra na figura 10.
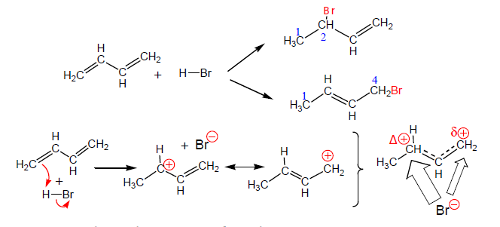
A reação inicia-se pela adição rápida de H+ à ligação dupla dando um carbocatião estabilizado por deslocalização eletrónica seguida do processo limitante da velocidade (ataque do anião brometo). A primeira estrutura contribuinte do carbocatião é mais importante do que a segunda por ter a carga localizada num carbono secundário, ou seja, o híbrido é mais parecido com esta estrutura do que com a segunda. À direita representa-se o híbrido com uma carga positiva maior no carbono 2 do que no carbono 4. Sendo assim, o ataque do brometo no carbono 2 é mais frequente do que o ataque no carbono 4, ou seja, o ataque no carbono 2 é mais rápido. A adição 1,2 é mais favorecida por questões cinéticas.
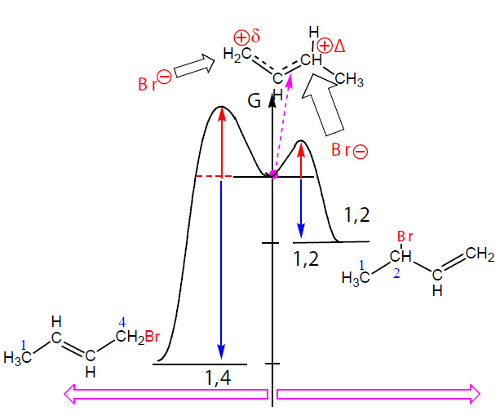
Sucede que os alcenos terminais, com a ligação dupla no início da cadeia, são menos estáveis termodinâmicamente que os alcenos com a ligação dupla no meio da cadeia, quer dizer, o aduto de adição 1,2, o que se forma mais rapidamente, é menos estável. Pelo contrário, o aduto 1,4, que se forma mais lentamente, é mais estável. O controlo da reação, cinético ou termodinâmico, depende da temperatura (tabela 1).

A temperaturas mais altas predomina o aduto 1,4, o mais estável; o controlo é termodinâmico. A temperaturas mais baixas predomina o adito 1,2, o que se forma mais rapidamente; o controlo é cinético. Jogando com a temperatura pode-se desviar a reação numa direção ou noutra.
Qual a razão da temperatura ser determinante no
tipo de controlo verificado?
A velocidade específica de um
processo elementar relacionase com a energia de ativação pela equação de
Arrhenius. Aplicando logaritmos teremos:
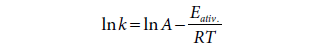
Se representarmos graficamente ln k em função de 1/T obtemos uma reta de coeficiente angular Eativ./R. Como a energia de ativação da reação que origina o aduto 1,4 é maior do que a produz o aduto 1,2, a reta correspondente a k1,4 é mais inclinada do que a que corresponde a k1,2 (Fig. 12).

A elevação da temperatura aumenta a velocidade de ambas as reações, favorece até mais a adição 1,4 (maior energia de ativação) em relação à adição 1,2. A cinética deixou, assim, de ser dominante, porque ambas as reações são rápidas, acabando por predominar o produto mais estável. O controlo é termodinâmico.
Se aquecermos a mistura de produtos obtidos a -80ºC (80% de 1,2 e 20% de 1,4) até 40ºC a mistura transformase em 20% de aduto 1,2 e 80% de aduto 1,4 mostrando a reversibilidade das reações. Assim, a temperaturas mais baixas o controlo é cinético; a temperaturas mais altas o controlo é termodinâmico.
A tabela 2 mostra outos exemplos.

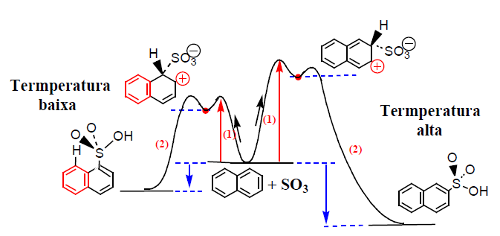
No caso do alceno, o mais estável é o aduto 1,4. No caso do naftaleno, o ácido 2-naftalenossulfonico é o mais estável por questões estereoquímicas. O ataque de SO3 na posição 1 do naftaleno é mais rápido devido a maior deslocalização eletrónica do aduto, ao qual o estado de transição se assemelha. Embora se possa escrever o mesmo número de estruturas contribuintes para os dois adutos, no ataque na posição 1 há duas estruturas que conservam a aromaticidade e no outro caso só há uma (Fig. 13).
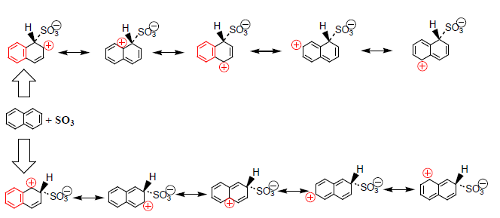
Na figura 14 pode ver-se que o passo limitante de velocidade é o ataque do anel (1), que é mais rápido quando se dá na posição 1. O segundo passo, (2), mais rápido e reversível, conduz aos ácidos sulfónicos correspondentes, sendo o ácido 2-naftalenossulfónico o mais estável por sofre menores repulsões estereoquímicas pelos átomos vizinhos.
Este artigo já foi visualizado 13751 vezes.