


Avogadro
Um programa de computador para editar e visualizar moléculas
📧
- Universidade do Porto
Referência Cerqueira, N., (2016) Avogadro, Rev. Ciência Elem., V4(2):017
DOI http://doi.org/10.24927/rce2016.017
Palavras-chave Molécula; 3D; visualização; Química;
Resumo
O Avogadro é um programa de computador muito versátil que permite construir, editar e visualizar moléculas em três dimensões. O programa pode ser utilizado para a compreensão de conceitos ligados à química tais como a geometria molecular, identificação dos ângulos de ligação, hibridizações, cálculo de energia e massa molecular, etc.
Este programa é compatível com vários sistemas operativos tais como o Windows, MacOS e Linux, podendo por isso ser instalado facilmente em qualquer computador. Para além disso, o programa é de licença livre, podendo ser distribuído livremente a qualquer pessoa.
O Avogadro pode ser utilizado para editar, manipular ou visualizar vários tipos de estruturas desde compostos orgânicos, nano-tubos de carbono, pequenas enzimas, etc.
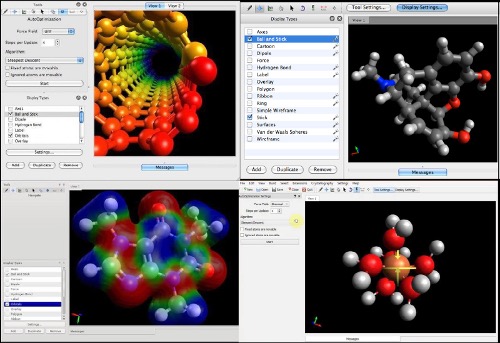
Na Figura 1 estão ilustradas algumas das potencialidades visuais do programa. Neste pequeno texto é feita uma breve abordagem sobre os principais comandos do Avogadro.
Funções básicas
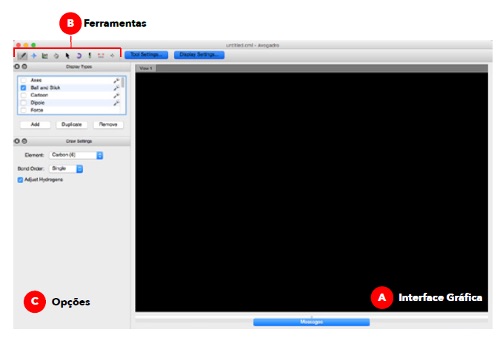
A interface do programa Avogadro é constituída por três secções (Figura 2). Na secção A ocorre a visualização e manipulação das moléculas. A secção B contém nove ferramentas diferentes que são utilizadas para manipular e editar as moléculas. Cada uma das ferramentas tem uma função especifica no programa. Sempre que uma ferramenta da secção B é selecionada, na secção C aparecem várias opções sobre essa ferramenta.
Nas secções seguintes é feita uma breve descrição sobre algumas das ferramentas da secção B e o seu modo de utilização.
 Modo de Edição
Modo de Edição
Ao selecionar o
ícone com o lápis, o Avogadro entra no seu modo de edição.
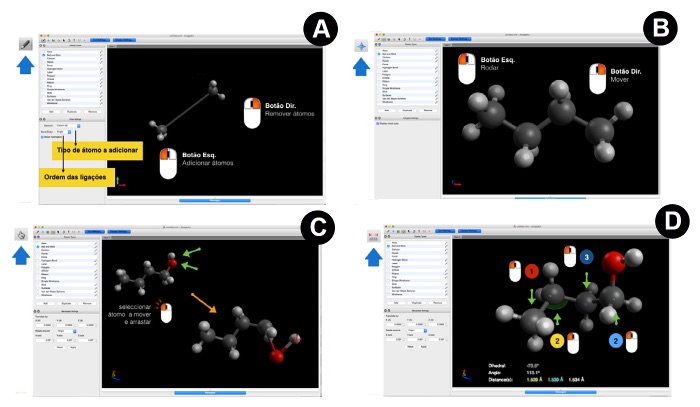
Para desenhar por exemplo um carbono, o utilizador deve carregar com o botão do lado esquerdo do rato em qualquer zona da secção A (Nota: o programa adiciona automaticamente os hidrogénio à molécula, por isso o resultado final será uma molécula de metano). Para adicionar outro átomo que esteja ligado ao átomo anterior basta carregar com o botão do lado esquerdo do rato sobre o átomo anterior e, mantendo o botão pressionado, arrastar o cursor para a posição onde se pretende colocar o novo átomo. No final obtém-se uma molécula de etano (Figura 3-A).
Para modificar a ordem da ligação entre dois átomos basta clicar com o botão do lado esquerdo do rato sob a ligação química. Dependendo do número de cliques, a ligação irá alternar entre uma ligação simples (etano), dupla (eteno) ou tripla (etino). Para adicionar outro tipo de átomo basta alterar o elemento na secção C. Na secção C também é possível alterar a representação dos átomos e das ligações químicas. Estão disponíveis vários estilos como esferas de Van der Waals, linhas (lines) , esferas e traços (ball and sticks), etc.
Para remover átomos na secção A basta clicar com o botão do lado direito do rato sobre o átomo que se pretendeeliminar. Caso os hidrogénios estejam a ser adicionados automaticamente, ao remover um átomo, eles também serão eliminados. Quando a construção da molécula estiver concluída, o passo seguinte envolve a otimização da sua geometria.
Para isso bastar ir ao menu Extensões e selecionar a opção Otimização de geometria. Durante este processo o Avogadro otimiza a geometria da molécula em função de uma energia que é calculada pelo programa. No final deste processo, a molécula adota uma conformação próxima de um dos seus mínimos de energia. Para gravar a molécula desenhada e visualizá-la mais tarde, basta ir ao menu FILE e gravar a estrutura com a opção SAVE. Existem várias formas de gravar um ficheiro contendo uma estrutura química, tais como o PDB, XYZ, MOL2, etc. É aconselhável a utilização da extensão MOL2 porque esta incluí a informação relativa à ordem das ligações químicas.
 Modo de
Visualização
Modo de
Visualização
Ao
selecionar o ícone com uma estrela azul, o Avogadro entra no modo de
visualização. Esta ferramenta permite mover, rodar, aproximar ou afastar
a molécula do campo de visão do utilizador. Para rodar a molécula basta
pressionar o botão do lado esquerdo do rato em qualquer área da secção A
e mover o rato ao mesmo tempo que o botão é pressionado. Para mover a
molécula deve-se repetir o mesmo procedimento anterior mas agora
pressionando o botão do lado direito do rato (Figura 3-B). Para ampliar
ou afastar a molécula deve-se usar o botão de scroll do rato.
 Modo de Manipulação
Modo de Manipulação
Ao selecionar o ícone com uma mão, o Avogadro entra no modo de manipulação.
Esta ferramenta é usada para alterar a posição de um determinado átomo na molécula. Isto poder ser útil, por exemplo, para alterar a conformação de um grupo de átomos.
Para alterar a posição de um átomo basta clicar com o botão do lado esquerdo do rato sobre o átomo que se pretende manipular na secção A. Depois mantém-se pressionado o mesmo botão do rato ao mesmo tempo que se arrasta o átomo para a nova posição. A nova posição do átomo será definida quando se largar o botão do lado esquerdo do rato (Figura 3-Cs).
Depois de se manipular os átomos pretendidos deve-se voltar a otimizar a geometria da molécula.
 Modo de Análise
Modo de Análise
Ao selecionar o
ícone com uma régua, o Avogadro entra no modo de análise, onde é
possível determinar comprimentos de ligação, ângulos e diedros. Para
calcular a distância entre dois átomos basta selecionar os átomos na
secção A com o botão do lado esquerdo do rato. O comprimento da ligação
calculado irá aparecer no canto inferior esquerdo da secção A. Para
determinar os ângulos e diedros, o mesmo procedimento deverá ser
utilizado, mas agora selecionado três e quatro átomos respetivamente. A
informação dos ângulos e diedros é apresentada no canto inferior
esquerdo da secção A (Figura 3-D).
 Modo de
Apresentação
Modo de
Apresentação
Ao
selecionar o ícone com uma seta em semi-círculo, o Avogadro entra no
modo de apresentação. Esta opção permite rodar da molécula representada
na secção A de forma automática. Para iniciar o modo de apresentação o
utilizador deve selecionar o grau de rotação em cada um dos seus eixos e
pressionar o botão START da secção C. Para parar o modo de apresentação
o utilizador deve pressionar o botão STOP.
Conclusão
O programa Avogadro
permite aos seus utilizadores ter um contacto mais estreito com as
moléculas. A sua utilização pode ser utilizada para ultrapassar
dificuldades de abstração e visualização que são normalmente
acompanhadas durante a aprendizagem deste tipo de conteúdos no ensino da
química.
Uma vez dominadas as funções básicas do programa, este poderá ser utilizado para visualizar e editar vários tipos de moléculas. Algumas bases de dados estão disponíveis na internet como por exemplo: http://www.chemspider.com.
Este software pode ser descarregado a partir
de:
http://avogadro.cc/wiki/Main_Page

Este artigo já foi visualizado 7510 vezes.