


Capacidades Caloríficas
📧
- Universidade do Porto
Referência Lage, E., (2019) Capacidades Caloríficas, Rev. Ciência Elem., V7(2):032
DOI http://doi.org/10.24927/rce2019.032
Palavras-chave temperatura; fluido; Termodinâmica; capacidade calorífica;
Resumo
A Capacidade Calorífica de um corpo é a quantidade de calor que é necessário fornecer-lhe para elevar a sua temperatura de uma unidade. Por outras palavras, é a quantidade de energia (na forma de calor) fornecida ao corpo dividida pela grandeza da variação da sua temperatura.
O conceito de capacidade calorífica é um dos mais importantes e mais antigos em Termodinâmica. A designação provém da velha teoria do calórico para a qual o calor era um fluido que passava de um corpo para outro e onde a quantidade trocada dependia das capacidades de armazenamento desse fluido que os corpos apresentariam. O calórico há muito foi abandonado, mas ficou o conceito de capacidade (\(C\)), a medida (unidade no S.I.: J/K) da quantidade de calor (\(\delta Q\)) que se tem de fornecer a um corpo para lhe originar uma elevação de temperatura \(\delta T\): \(C=\frac{\delta Q}{\delta T}\), i.e., quantos Joules são necessários para elevar de 1K a temperatura da substância. Alguns exemplos, referidos a 1Kg: aço (460), granito (800), vidro (40), água (4184). Quando referida a 1 Kg, a capacidade calorífica designa-se por calor específico ou capacidade calorífica mássica. Note-se que, para a água, este resultado é exato porque é a definição da Kcal (1000 calorias). Esta definição é, contudo, insuficiente por ser necessário especificar as condições a que o sistema está sujeito quando recebe calor, sendo comum ser mantido constante o volume (\(V\)) ou a pressão (\(p\)), assim se definindo a capacidade calorífica a volume constante (\(C_V\)) ou pressão constante (\(C_p\)), respetivamente. Iremos aqui considerar a relação entre estas capacidades caloríficas e expôr o método de Rüchardt que determina experimentalmente \(\gamma \equiv C_p/C_V\) em gases.
Para os sistemas simples que aqui consideramos, o estado de equilíbrio termodinâmico pode ser definido pelo volume (\(V\)) e temperatura (\(T\)), deduzindo-se a pressão a que está submetido pela equação de estado que relaciona estas três variáveis. Assim, a entropia, uma função de estado, tem \(V\) e \(T\) como argumentos, pelo que a sua diferencial se escreve:
\(dS=\left(\frac{\partial S}{\partial T}\right)_VdT + \left(\frac{\partial S}{\partial V}\right)_TdV\) (1)
Aqui, os subscritos indicam a variável que é mantida fixa na derivação.
Comecemos por analisar o caso simples de ser mantido constante o volume. Então, quando o sistema recebe, reversivelmente, a quantidade de calor \(\delta Q\), a sua entropia aumenta de \(\frac{\partial Q}{T}\), pelo que:
\(dS=\frac{\delta Q}{T}=\left(\frac{\partial S}{\partial T}\right)_VdT \ \ \ \ \ \ \ \Rightarrow \ \ \ \ \ \ \ C_V=\frac{\delta Q}{\delta T}=T\left(\frac{\partial S}{\partial T}\right)_V\) (2)
Suponhamos, agora, que é mantida constante a pressão. Então, a quantidade de calor que o sistema necessita receber para elevar a sua temperatura de \(\delta Q\) é maior que no caso anterior porque parte desse calor é transformado em trabalho pois que, necessariamente, há aumento de volume (dilatação térmica) para ser mantida a pressão. Assim, deverá ser e \(C_p\geq C_V\), usando a eq. (1) podemos relacionar estas duas capacidades:
\(dS=\frac{C_V}{T} dt +\left(\frac{\partial S}{\partial V}\right)_T\left(\frac{\partial V}{\partial T}\right)_p dt\)
Segue-se que a quantidade de calor recebida é:
\(\delta Q =TdS=\left[C_V+T\left(\frac{\partial S}{\partial V}\right)_T\left(\frac{\partial V}{\partial T}\right)_p\right]dT \ \ \ \ \ \ \ \Rightarrow \ \ \ \ \ \ \ C_p=C_V+T\left(\frac{\partial S}{\partial V}\right)_T\left(\frac{\partial V}{\partial T}\right)_p\) (3)
A equação de estado fornece \(\left(\frac{\partial V}{\partial T}\right)_p\)- abaixo veremos o resultado para um gás perfeito. Mas como calcular \(\left(\frac{\partial S}{\partial V}\right)_T\)? Entra, aqui, todo o poder analítico da Termodinâmica: devemos procurar uma função de estado cuja diferencial se escreve à custa das diferenciais do volume e da temperatura. Essa função de estado não é a energia interna (\(U\)) pois que os 1º e 2º Princípios da Termodinâmica nos levam a escrever:
\(dU=TdS-pdV\)
Mas esta expressão sugere a forma de encontrarmos o que procuramos 1: considere-se a função \(F\equiv U-TS\), conhecida por energia livre de Helmholtz. Então:
\(dF=dU-TdS-SdT=-SdT-pdV\) (4)
Esta diferencial é exata porque \(F\) é uma função de estado, pelo que:
\(\frac{\partial }{\partial V}\left(\frac{\partial F}{\partial T}\right)_V=\frac{\partial }{\partial T}\left(\frac{\partial F}{\partial V}\right)_T\)
Ora, da eq. (4), obtemos:
\(\left(\frac{\partial F}{\partial T}\right)_V=-S \ \ \ \ \ \ \ \ \left(\frac{\partial F}{\partial V}\right)_T=-p\)
Pelo que a relação anterior fica 2:
\(\left(\frac{\partial S}{\partial V}\right)_T=\left(\frac{\partial p}{\partial T}\right)_V\)
O segundo membro é conhecido pela equação de estado. Assim, regressando à eq. (3), obtemos:
\(C_p=C_V+T\left(\frac{\partial p}{\partial T}\right)_V\left(\frac{\partial V}{\partial T}\right)_p\) (5)
Esta relação aplica-se a qualquer corpo. Num sólido, por exemplo, a dilatação térmica é muito pequena e pode, muitas vezes, ser ignorada, pelo que \(C_p\approx C_V\). Não é assim nos gases, como veremos adiante.
Ex.: gás perfeito
A equação de estado é \(p=\frac{nRT}{V}\), onde \(n\) é o número de moles e \(R\) a constante dos gases perfeitos. Então:
\(\left(\frac{\partial p}{\partial T}\right)_V=\frac{nR}{V} \ \ \ \ \ \ \ \ \left(\frac{\partial V}{\partial T}\right)_p=\frac{nR}{p}\ \ \ \ \ \ \Rightarrow \ \ \ \ \ \ T \left(\frac{\partial p}{\partial T}\right)_V\left(\frac{\partial V}{\partial T}\right)_p=T\frac{nR}{V}\frac{nR}{p}=nR\)
Assim:
\(C_p=C_V+nR\) (6)
Que podemos dizer sobre \(C_V\)? O teorema da equipartição da energia informa que cada termo quadrático na energia de uma molécula contribui com \(\frac{1}{2}\frac{R}{N_A}T\) para a energia média da molécula (esta não tem uma energia fixa quando em contacto com a fonte de calor à temperatura \(T\)). Então, para as \(nN_A\) moléculas, cada termo quadrático contribui com \(\frac{1}{2}nRT\). Se houver \(f\) termos quadráticos na energia da molécula, e admitindo que as moléculas não interagem (gás perfeito!), segue-se que \(U=f\frac{1}{2}nRT\), pelo que \(C_V=f\frac{1}{2}nR\). Alguns exemplos: num gás monoatómico, é \(f = 3\) porque cada átomo só contribui com a energia cinética do movimento, havendo, pois, 3 termos quadráticos correspondentes às 3 componentes da velocidade. Num gás diatómico, para além da velocidade do centro de massa (contribuindo com os mesmos 3 termos para a energia cinética), existem, agora, 2 termos adicionais associados a movimentos de rotação em torno de 2 eixos perpendiculares entre si e perpendiculares à linha que une os átomos. Assim, \(f = 5\) se a molécula se comportar como um haltere rígido. Mas se admitirmos que a molécula pode vibrar como um oscilador harmónico, surgem mais 2 termos quadráticos originados nas energias cinética e potencial da “mola” que une os átomos, pelo que \(f = 7\). No caso geral, somos, assim, conduzidos, pela eq. (6), à expressão:
\(C_p=\frac{f}{2}nR+nR=\left(\frac{f}{2}+1\right)nR \ \ \ \ \ \ \ \ \ \ \Rightarrow \ \ \ \ \ \ \ \ \gamma\equiv\frac{C_p}{C_v}=\frac{f+2}{f}\)
A tabela seguinte mostra os valores esperados para diversos modelos.
| \(f\) | \(γ\) | Modelo da molécula |
| 3 | 5/3 | monoatómico |
| 5 | 7/5 | haltere |
| 7 | 9/7 | Diatómica c/vibração |
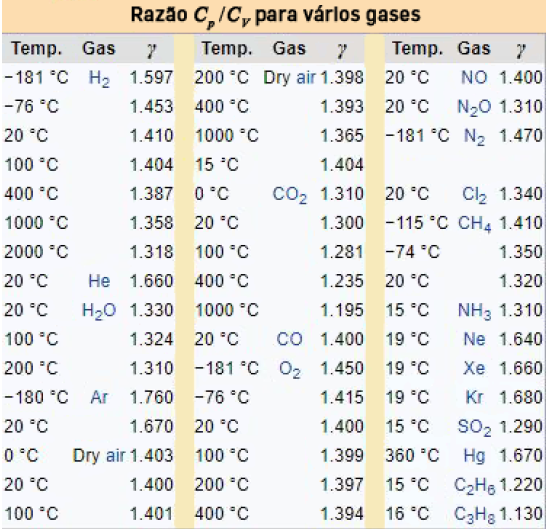
A tabela abaixo mostra valores de \(γ\) obtidos experimentalmente para vários gases. Os resultados deduzidos para um gás perfeito não nos devem iludir: em geral, as capacidades caloríficas variam com a temperatura, fornecendo, desse modo, importante informação sobre o “congelamento” de graus de liberdade moleculares quando, por exemplo, a temperatura diminui. Atente-se no caso da molécula de hidrogénio: acima de 100 ºC, os valores de \(γ\) sugerem um modelo haltere, mas a -181 ºC, já a molécula parece não poder rodar!
Um último exemplo considera um modelo simples de um sólido: cada átomo está ligado à sua posição de equilíbrio por uma “mola” (potencial harmónico), o que dá \(f = 6\) (3 termos quadráticos na energia cinética e outros 3 termos quadráticos na energia potencial). Assim, para um mole de um sólido será de esperar \(C_V ≃ 3R\), o que parece reproduzir a lei de Dulong e Petit (1819), mas apenas para temperaturas elevadas. Contudo, quando se diminui a temperatura, a capacidade calorífica molar diminui rapidamente porque é necessário considerar a quantificação destes osciladores harmónicos, como Einstein mostrou em 1907. Este é um importante tópico que aqui não será mais desenvolvido.
Voltando aos gases, admitamos que as capacidades caloríficas são constantes, independentes da temperatura. Nestas condições, podemos deduzir a equação das adiabáticas, i.e., a relação entre pressão e volume quando o sistema evolui mantendo a entropia constante. De facto, regressando à eq. (1), e com os resultados já obtidos, podemos escrever:
\(dS=\frac{C_V}{T}dT+\left(\frac{\partial p}{\partial T}\right)_V dV=\frac{C_V}{T}dT+\frac{nR}{V}dV=0\)
A equação integra-se facilmente, obtendo-se:
\(TV^{\frac{nR}{C_v}}=const\)
Ou, eliminando a temperatura pela equação de estado, tem-se a forma mais familiar:
\(pV^{1+\frac{nR}{C_V}}=pV^\gamma=const\) (7)
Um método experimental para a determinação direta de \(γ\) foi apresentado por Eduard Rüchardt (FIGURA 1), em 1928.

O dispositivo (FIGURA 2) consiste num grande balão onde se introduz o gás a investigar. O balão liga ao exterior através de uma longa coluna cilíndrica de vidro (diâmetro interno \(2R\)) colocada verticalmente. Uma esfera (ou um cilindro) maciça, de aço, de massa \(m\) e com um raio muito ligeiramente inferior ao do tubo, é introduzida neste.
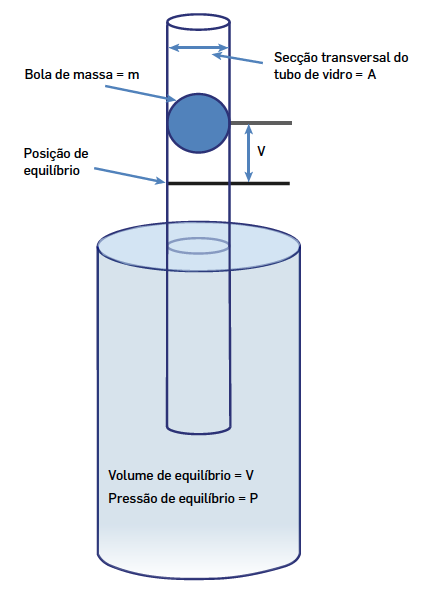
Ignorando o atrito existe uma posição de equilíbrio para a esfera quando a força devida à pressão do gás no balão igualar a soma do peso da esfera com a força devida pela pressão atmosférica. Tomando o eixo \(z\) vertical e designando por \(z = 0\) a cota desta posição de equilíbrio, então deve ser:
\(\pi R^2p(0)=mg+\pi R^2 p_a\)
Que acontece quando a esfera é afastada desta posição? Se a esfera descer, comprime o gás dentro do balão, aumentando a pressão que ele exerce e, portanto, a esfera será empurrada para \(z = 0\); se a esfera subir, aumenta o volume ocupado pelo gás, o que diminui a pressão que ele exerce e, portanto, a esfera será puxada para \(z = 0\). Assim, se a esfera for largada verticalmente no interior do tubo, ficará a oscilar, podendo medir-se o período da oscilação, deduzindo-se a partir daí o valor de \(γ\). Com efeito, as compressões e expansões do gás são tão rápidas que o gás, praticamente, não troca calor com o exterior, i.e., evolui adiabaticamente. Assim, designando por \(p(z)\) e \(V(z)\), respetivamente, a pressão exercida pelo gás e o volume que ocupa quando a esfera está à cota \(z\), tem-se, pela eq. (7).
\(p(z)V(z)^\gamma =p(0)V(0)^\gamma \ \ \ \ \Leftrightarrow \ \ \ \ p(z)=p(0)\left[\frac{V(0)}{V(z)}\right]^\gamma\)
Ora, \(V(z)=V(0)+\pi R^2z\), pelo que:
\(p(z)=p(0)\left[1+\frac{\pi R^2z}{V(0)}\right]^{-\gamma}\simeq p(0)\left[1-\gamma\frac{\pi R^2z}{V(0)}\right]\)
onde se admitiu \(V(0)>>\pi R^2z\) porque o balão é grande e as oscilações têm pequena amplitude. Assim, as forças que atuam na esfera têm resultante vertical
\(\left[p(z)-p_a\right]\pi R^2-mg=\left[p(z)-p(0)\right]\pi R^2\cong-p(0)\gamma\frac{(\pi R^2)^2}{V(0)}z\)
tendo, a primeira igualdade sido obtida por eliminação do peso através da condição de equilíbrio. Assim, a lei fundamental da dinâmica dá, para o movimento da esfera:
\(m\frac{d^2z}{dt^2}=-p(0)\gamma\frac{(\pi R^2)^2}{V(0)}z\)
É a equação de um oscilador harmónico que, portanto, apresenta a frequência (angular):
\(\omega= \sqrt{p(0)\gamma \frac{(\pi R^2)^2}{mV(0)}}\)
Com \(p(0)\) determinado pela posição de equilíbrio, \(V(0)\) praticamente igual ao volume do balão, obtemos assim \(γ\) medindo o período \(\frac{2\pi}{\omega}\) das oscilações da esfera.
Notas
1 A passagem de \(U\) para \(F\) é uma transformação de Legendre; o uso destes
“truques” tornam a Termodinâmica algo abstrata, mas simultaneamente, muito poderosa.
2 O resultado obtém-se diretamente da eq. (4): as derivadas cruzadas são iguais.
Este artigo já foi visualizado 10673 vezes.