


Bases de Dados de Proteínas
📧
- LAQV/ REQUIMTE/ DQB/ FCUP
Referência Oliveira, A., (2022) Bases de Dados de Proteínas, Rev. Ciência Elem., V10(4):052
DOI http://doi.org/10.24927/rce2022.052
Palavras-chave Proteína, proteómica - bioinformática, estrutura primária, estrutura secundária, estrutura tridimensional, Função
Resumo
A biologia, a química e a bioquímica têm-se vindo a transformar em ciências ricas em dados. Atualmente, geram-se enormes quantidades de dados para estruturas tridimensionais, dados de atividade e funções, e particularmente sobre a estrutura primária de proteínas. Com o aumento de dados, cresceu também a necessidade de armazenar, integrar e comunicar estes grandes conjuntos de informação proteómica como um todo. Neste pequeno texto, focamo-nos na UniProt, a base de dados universal de proteínas, que reúne em si informação de várias outras bases de dados e, por isso, permite aos investigadores o acesso rápido e fácil a quantidades de informação massiva.
A necessidade de uma Base de Dados em Proteínas
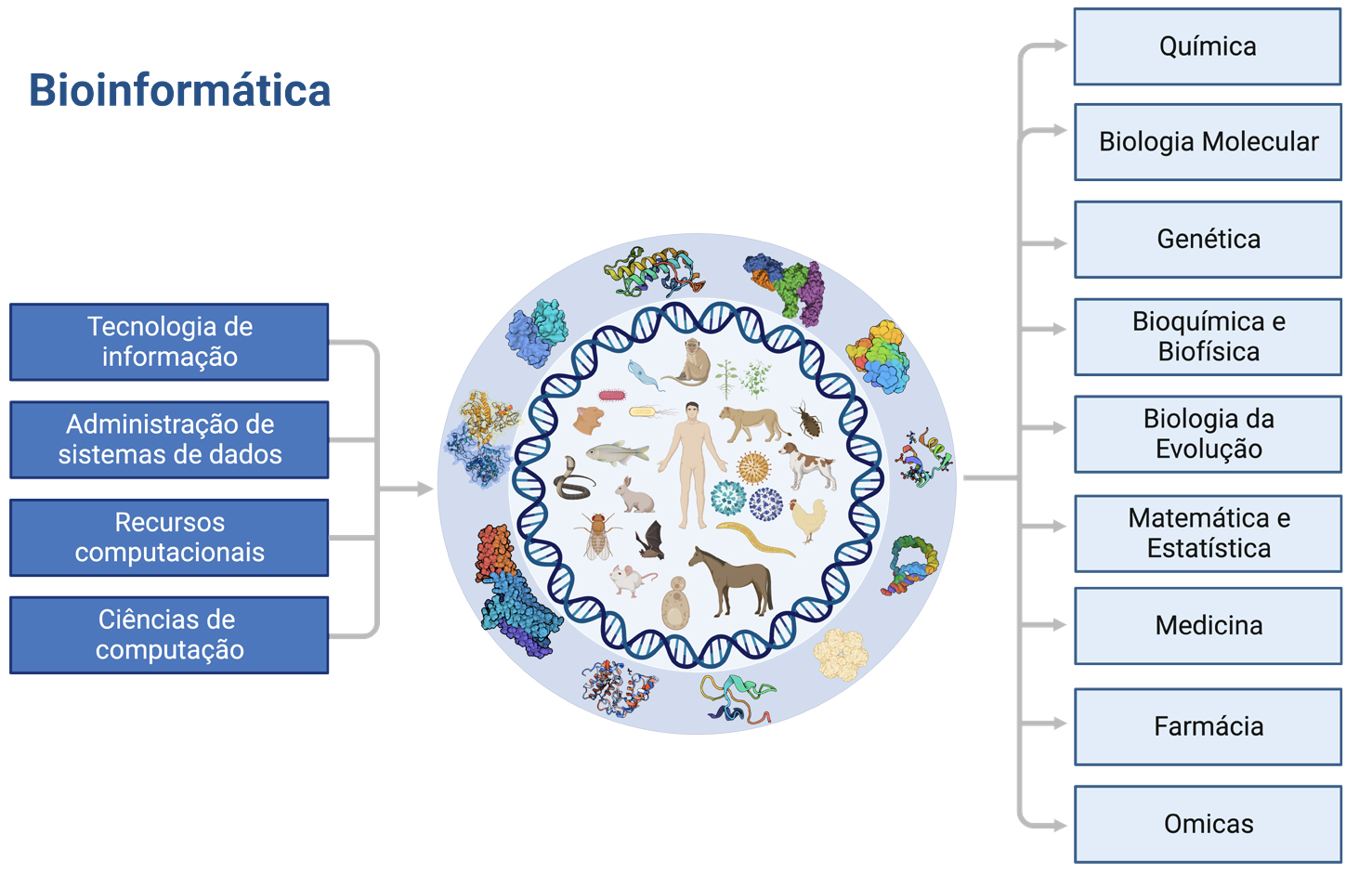
Não podemos falar em bases de dados sem primeiro introduzir, brevemente, a Bioinformática. Esta emergente área interdisciplinar combina o poder computacional e técnicas informáticas, matemáticas e de estatística com a biologia, a química, a farmácia e a medicina (FIGURA 1). Margaret O. Dayfoff foi, sem dúvida, a mãe da bioinformática e a pioneira no desenvolvimento das suas técnicas. Foi ela quem pela primeira vez aplicou métodos matemáticos e computacionais à bioquímica, criando as primeiras bases de dados de proteínas e ácidos nucleicos. Dayoff deu origem ao atual código de uma letra dos aminoácidos, numa tentativa de reduzir o volume de dados, para que os (super) computadores da época pudessem armazenar mais informação. Desde então, a informática sofreu grandes avanços e a capacidade de armazenamento de dados aumentou exponencialmente. Em paralelo, ocorreram grandes avanços nas ciências genómicas e nas tecnologias de sequenciação de próxima geração, as quais permitiram, nas últimas décadas, descobrir informação genómica e proteómica de um variado número de organismos. Como consequência, o número de proteínas sequenciadas e arquivadas em bases de dados tem aumentado drasticamente nos últimos anos.
As proteínas são macromoléculas de particular interesse porque ocupam o campo molecular intermédio entre o gene e a transcrição, e desempenham um papel fundamental na estrutura e organização molecular e celular. Além disso, a maioria dos processos fisiológicos e patológicos manifesta-se a nível proteico, e, portanto, torna-se emergente a utilização de técnicas proteómicas e bioinformáticas de alto rendimento para alcançar uma melhor compreensão da biologia molecular básica e das suas alterações em casos de doenças. A comparação entre proteínas, ou entre famílias de proteínas, fornece informação sobre a sua relação no genoma ou com outras proteínas relativas em outras espécies, e também permite a identificação do porquê da sua implicação em doenças. Assim, a integração de dados sobre várias proteínas permite obter muito mais informação que aquela dada por uma simples proteína isolada.
A riqueza dos novos dados proteómicos permite aos investigadores formular perguntas biológicas complexas e obter novos conhecimentos científicos que suportem estas novas hipóteses orientadas por dados. Nos últimos anos, foram desenvolvidas muitas bases de dados bioinformáticas relacionadas com proteínas, instalações de consulta e ferramentas de software de análise de dados para organizar e fornecer anotações biológicas de proteínas que apoiem análises sequenciais, estruturais, funcionais e evolutivas no contexto da bioquímica de redes e sistemas.
A evolução de bases de dados de proteínas
A necessidade de reunir informação sobre a sequência de proteínas remonta a 1965, quando se publica o Atlas da Sequência e Estrutura de Proteínas, editado por Margaret O. Dayhoff. Este trabalho reunia apenas a informação sequencial de 66 proteínas diferentes, agrupadas em 10 categorias e continha aspetos muito básicos como a formação de pontes persulfureto ou os locais de interação de cofatores. Nesta altura procurava-se já relacionar a sequência da proteína com a sua estrutura e obter pequenas conclusões através da comparação com proteínas relativas.
Depois, em 1971, surge o Banco de dados de Proteínas (PDB)a, um repositório público e livre focado na biologia estrutural. O PDB contém toda a informação conhecida sobre a estrutura tridimensional de péptidos, proteínas, ácidos nucleicos e complexos formados entre macromoléculas biológicas ou entre macromoléculas e pequenos compostos orgânicos ou mesmo fármacos. Atualmente, contém mais de 198 mil estruturas determinadas experimentalmente por técnicas de raios-X, NMR e microscopia eletrónica, e cerca de 1 milhão de modelos obtidos computacionalmente por técnicas de bioinformática.
Em 1984, o atlas de sequência de proteínas deu lugar à primeira base de dados mundial de sequências de proteínas classificadas e funcionalmente anotadas — PIR-PSD. O PIR-PSD classifica as sequências de proteínas com base no conceito de superfamília. Estas sequências são também classificadas com base no domínio da homologia e nos motivos da sequência, ou seja, os domínios de homologia podem corresponder a blocos de construção evolutivos, enquanto os motivos de sequência representam sítios funcionais ou regiões conservadas. Esta abordagem na classificação permite uma compreensão mais completa da relação função-estrutura da sequência e da sua evolução.
Mais tarde, em 1986, nasce, na Universidade de Genebra, a primeira base de dados que visa reunir a informação detalhada sobre uma dada proteína — a SWISS-PROT DB. Para cada proteína incluída nesta base de dados estão disponíveis três tipos de informação: (i) a sequência da proteína; (ii) informação bibliográfica; e (iii) a sua informação taxonómica. Sempre que possível inclui-se também informação sobre a função da proteína, isoformas, as modificações pós-tradução, informação detalhada sobre a estrutura como os domínios e resíduos catalíticos, mutações, a estrutura secundária e quaternária, similaridade com outras proteínas, doenças relacionadas e conflitos na sequência.
Finalmente, em 2002, forma-se o consórcio da UniProt. Este reúne equipas de investigação do Instituto Europeu de Bioinformática (EBI), do Instituto Suíço de Bioinformática (SIB) e do Recurso de Informação sobre Proteínas (PIR).
UniProt
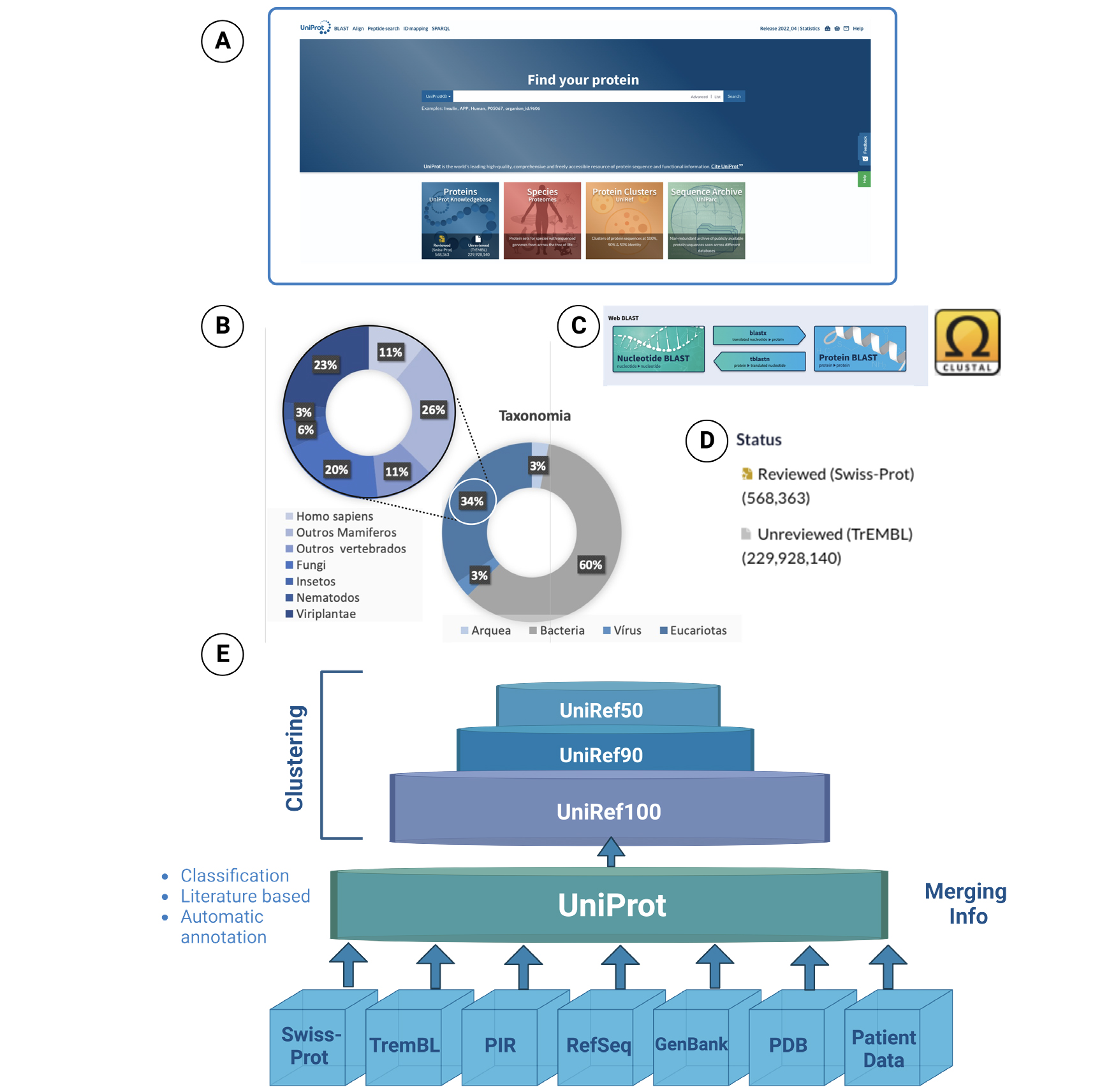
A UniProtb, ou base universal de proteínas, é a maior base de dados mundial com informação sobre proteínas. Continha, em Novembro de 2022, cerca de 289 milhões de sequências únicas, que incluem sobretudo proteínas de bactérias e organismos eucariotas, e quase 283 mil proteomas distintosc, compreendendo mais de 205 milhões de combinações de aminoácidos. Esta base de dados abrangente e não redundante de sequências de proteínas arquivadas, reúne a informação disponível em todos os principais recursos acessíveis ao público como a Swiss-Prot, a PIR e o PDB, referidos acima, e também a informação disponibilizada em outras bases de dados como o TremBLd, RefSeqe, GenBankf e bases de dados médicas com informação sobre pacientes. A cada sequência de proteínas, ou entrada em linguagem informática, é atribuído um código identificador único e um nome que identifica cada proteína tendo em conta o gene que a codifica e o organismo de que provém. Por exemplo, a ciclooxigenase-2 (COX2) humana, uma enzima extremamente estudada pelas suas implicações em processos inflamatórios, tem o código identificador P35354 e o nome PGH2_HUMAN. Se o leitor pretender procurar todas as sequências de cicloxigenase-2 conhecidas, até à data, bastaria aceder à página web da base de dados e no local de busca (Find your protein), identificado por (A) na FIGURA 2, escrever cyclooxygenase 2. Em alguns segundos, a base de dados encontra 17 235 resultados, dos quais 17 200 são sequências consideradas não revistas e apenas 35 são entradas revistas. Mas então que diferença há entre a informação nas entradas? As sequências que tiveram a sua origem na base de dados de SWISS-PROT (cerca de 570 mil no total) são consideradas entradas revistas porque contêm informação validada por especialistas, que para além da informação da estrutura primária das proteínas, inclui também isoformas e variantes, informação detalhada sobre a sua função, mecanismo enzimático (quando aplicável), estrutura tridimensional, interações com outras proteínas ou DNA e a sua implicação em vias metabólicas e em doenças. Finalmente, e como já foi referido anteriormente, para uma melhor compreensão da informação, não devemos ver as proteínas isoladas, mas agrupadas com base na sua similaridade e identidade sequencial. Os bioinformáticos desenvolveram, por isso, técnicas de comparação da identidade estrutural de proteínas. Não poderíamos aqui focar-nos em todas as técnicas existentes, mas destacamos duas das mais importantes: Basic Local Alignment Search Tool (BLAST)g e CLUSTALh, implementadas na base de dados. A primeira procura regiões de proteínas com sequência semelhante, enquanto a segunda compara as semelhanças entre 2 ou mais sequências. Assim, na UniProt, sempre que possível, as sequências das proteínas são agrupadas, em três grupos distintos: 100% UniRef100 (se as proteínas compartem 100% da sua identidade sequencial); UniRef90 (quando a identidade sequencial \(\geq \) 90%) ou UniRef50 (identidade \(\geq \) 50%). A cada um destes grupos é atribuído um representante, isto é, a sequência proteica do grupo melhor caracterizada, até à data, um grupo de proteínas relacionadas.
Um exemplo prático na sala de aula
Atualmente, a pesquisa em bases de dados é, com frequência, o primeiro passo no estudo de uma nova proteína. A utilização de bases de dados científicas ao nível do ensino básico e secundário, permite ao estudante realizar uma investigação autêntica, de forma análoga a como muitos cientistas levam a cabo, hoje em dia, a sua investigação. Neste contexto apresentamos aqui um exemplo prático da utilização da UniProt (FIGURA 3).
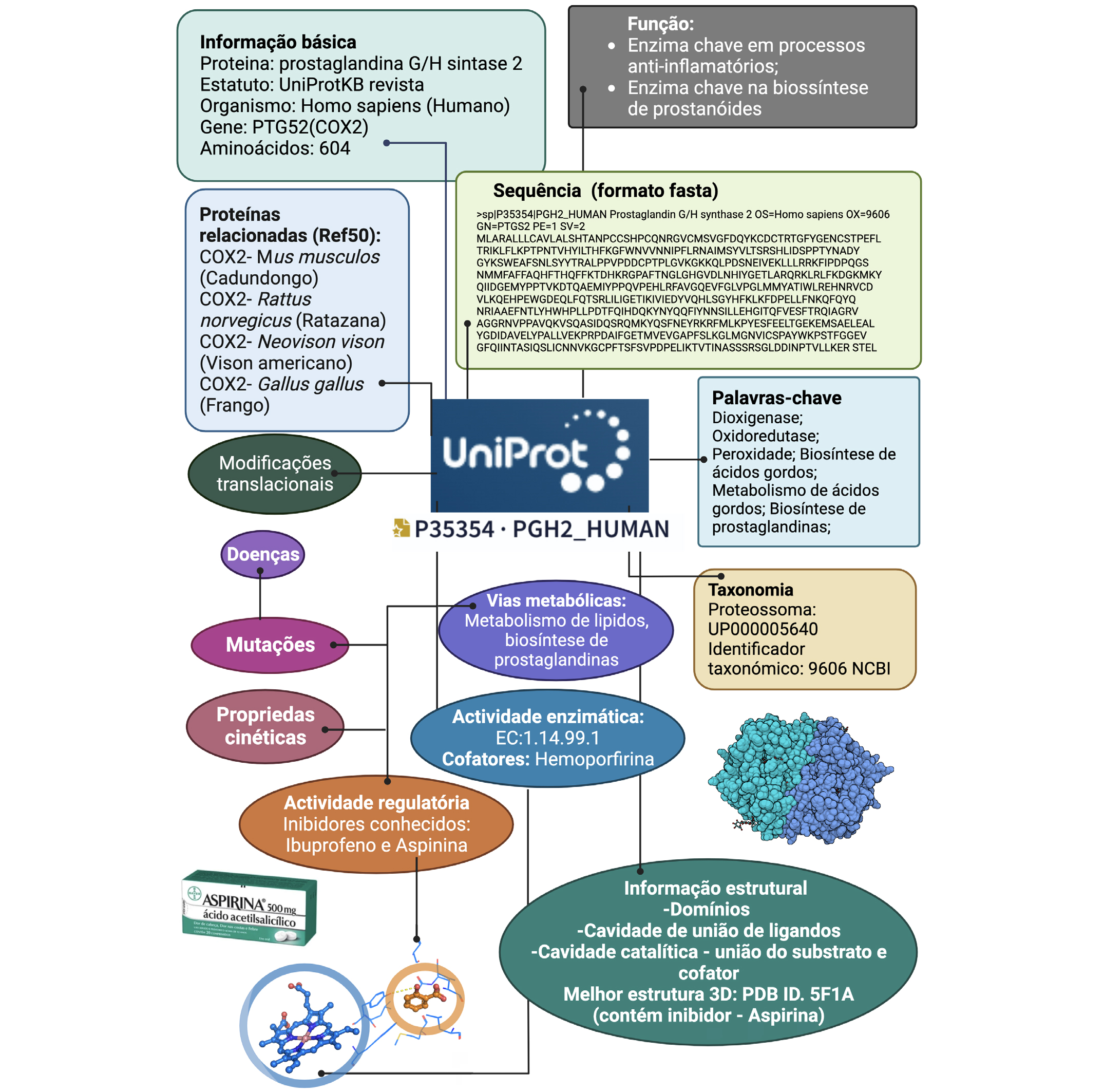
Vejamos então em detalhe a informação contida no exemplo acima, a entrada P35354. Ao abrirmos o enlace associado ao código identificador, de imediato, encontramos o nome detalhado da proteína: Protaglandin G/H synthase 2, o organismo a que pertence Homo sapiens (o qual tem associado um identificador taxonómico e de proteossoma) e o nome do gene que a codifica PTGS2(COX2). No campo Função descreve-se, sumariamente, o papel que esta proteína tem a nível molecular e as vias metabólicas em que está implicada, assim como as referências na literatura que suportam os dados (códigos PUBMED). Depois, encontramos um campo de informação variada, onde, entre outra informação, destacamos que esta proteína está identificada como alvo de medicamentos anti-inflamatórios, como o Brufen® e a Aspirina®. Segue-se uma descrição detalhada do seu mecanismo químico, onde constatamos que esta enzima converte, na presença de hemoporfirina (cofator), ácido araquidónico em prostaglandinas, implicados em processos de inflamação e percebemos o porquê desta enzima unir anti-inflamatórios, e consequentemente, a sua inatividade causada pelos fármacos já descritos. Finalmente, destacam-se dois aspetos: os aspetos estruturais, ou seja, a existência de estrutura tridimensional conhecida e depositada no PDB, onde se identificam as regiões relevantes da proteína, que são essenciais para a sua função, como é o caso da hemoporfirina, locais de união de substratos e inibidores, pontes persulfureto, etc; e os aspectos de similaridade com outras proteínas, onde de demonstra que apenas quatro proteínas são semelhantes sequencialmente à COX-2 humana, e estas são a COX2 presente nos organismos: o camundongo, a ratazana, o vison americano e o frangoi.
Notas
c O UniProt Proteomes fornece conjuntos de proteínas que são consideradas como sendo expressas por organismos cujos genomas foram completamente sequenciados.
d TrEMBL (do ingles “Translated EMBL”) é uma base de dados de sequências de proteínas anotadas por computador que é divulgada como um suplemento da SWISS-PROT. Contém a tradução de todas as sequências de codificação presentes na base de dados do EMBL Nucleotide, que não foram totalmente anotadas.
e RefSeq-NCBI fornece sequências curadas não redundantes de regiões genómicas, transcrições e proteínas. Inclui regiões de codificação, domínios conservados, variações, etc. e anotações melhoradas, tais como publicações, IDs Gene, e referências cruzadas de bases de dados.
f GenBank-NIH é uma base de dados de sequências genéticas, criada para melhor compreensão da informação contida no DNA, isto é, contem as sequencias dos nucleótidos que codificam a informação de proteínas.
g https://blast.ncbi.nlm.nih.gov/Blast.cgi.
h https://www.ebi.ac.uk/Tools/msa/clustalo/.
i Para outras utilizações mais extensivas de UniProt na sala de aula recomendamos o uso do tutorial desenvolvido por Michele Magrane, EMBL-EBI: https://www.ebi.ac.uk/sites/ebi.ac.uk/files/content.ebi.ac.uk/materials/2011/111006_SME/uniprot_tutorial_-_michele_ magrane.pdf.
Referências
- 1 VAN DER GAAF, W., Contributions to the history of english, Neophilologus, 12. 1927.
- 2 MASIC, I., The most influential scientists in the development of medical informatics (13): Margaret Belle Dayhoff, Acta Inform Medica, 24, 299. 2016.
- 3 DAYHOFF, M. O. et al., Atlas of protein sequence and structure, National Biomedical Research Foundation, 185. 1978.
- 4 BERMAN, H. et al., Announcing the worldwide Protein Data Bank, Nat. Struct. Biol., 10, 980. 2003.
- 5 WU, C. H. et al., The iProClass integrated database for protein functional analysis, Comput. Biol. Chem., 28, 87–96. 2004.
- 6 O’DONOVAN, C. et al., High-quality protein knowledge resource: SWISS-PROT and TrEMBL, Brief. Bioinform, 3, 275– 284. 2002.
- 7 LEINONEN, R. et al., UniProt archive, Bioinformatics, 20, 3236–3237. 2004.
- 8 FAMIGLIETTI, M. L. et al., An enhanced workflow for variant interpretation in UniProtKB/Swiss-Prot improves consistency and reuse in ClinVar, Database, 2019, 1–8. 2019.
- 9 BATEMAN, A. et al., UniProt: the universal protein knowledgebase in 2021, Nucleic Acids Res., 49, 480–489. 2021.
- 10 COUDERT, E. et al., Annotation of biologically relevant ligands in UniProtKB using ChEBI, 1 Introduction 2 Methods, 1–6. 2022. DOI: 10.1093/bioinformatics/xxxxx.
Este artigo já foi visualizado 5887 vezes.