


CRISPR/Cas
Definição, aplicações e desafios na biotecnologia vegetal
📧 , 📧 , 📧 , 📧
- * CEF/ LAT/ DCV/ U. Coimbra
- ɫ DB/ U. Porto
- ‡ CEF/ LAT/ DCV/ U. Coimbra
- + CEF/ LAT/ DCV/ U. Coimbra
Referência Ferraz, R., Coimbra, S., Correia, S., Canhoto, J., (2023) CRISPR/Cas, Rev. Ciência Elem., V11(1):009
DOI http://doi.org/10.24927/rce2023.009
Palavras-chave edição genética, endonucleases, quebras na cadeia dupla de DNA (DSB), recombinação genética, RNA-guia
Resumo
As técnicas de modificação de genomas têm como objetivo a alteração de uma sequência específica de DNA. Com o evoluir destas técnicas foi possível desenvolver sistemas que envolvem enzimas reprogramáveis que estão no cerne das técnicas de edição genética atuais. Entre estas novas abordagens, um novo sistema viria revolucionar o mundo da engenharia genética, a técnica CRISPR/Cas. Esta técnica apresenta diversas aplicações na edição de genomas de espécies vegetais, tendo sido utilizada para melhorar diversas características, como a produtividade e a gestão de stresse, em plantas com interesse económico como o tomateiro. Embora esta nova abordagem apresente alguns pormenores que podem ainda ser melhorados, a potencialidade que apresenta para a modificação de genomas torná-la-á muito importante no melhoramento de plantas num futuro próximo.
As tecnologias de modificação de genomas têm como objetivo a alteração de uma sequência específica de DNA. A edição genética com fins biotecnológicos começou a ser aplicada nos anos 70 do século XX, com o estudo da recombinação homóloga, processo no qual uma sequência de DNA ladeada por regiões homólogas do DNA hospedeiro pode ser integrada no genoma deste1. No entanto, a utilização direta desta técnica apresenta várias limitações, como a baixa eficácia de recombinação e a integração aleatória em zonas do genoma não desejadas2. Para além disso, embora a utilização de organismos geneticamente modificados tenha um forte e positivo impacto no melhoramento de espécies vegetais com interesse económico, estas tecnologias não têm uma aceitação pública generalizada, em especial nos países europeus, o que tem dificultado a sua adoção3, 4. A aplicação da recombinação homóloga no melhoramento de plantas envolve a adição de material genético numa planta de interesse, modificando assim o seu genoma, sendo que o material genético adicionado pode ser da mesma planta ou ser estranho à planta a ser modificada, que, neste último caso, passa a ser designada de planta transgénica5. A baixa frequência de integração num local específico desejado e a necessidade de utilizar genes marcadores e/ou de seleção limitam bastante a utilização das técnicas de recombinação genética6.
Com a evolução das técnicas de edição genética, foi possível desenvolver novos sistemas que não envolvem as antigas enzimas de recombinação, mas sim novas enzimas de restrição e nucleases reprogramáveis que estão no cerne das técnicas de edição genética atuais7. Para além disso, foi também crucial a associação entre as quebras na dupla cadeia do DNA (Double-Strand Breaks, DSB) e uma maior integração de DNA estranho no material genético de células de mamíferos6, 8. A descoberta de meganucleases, enzimas de restrição com sequências de reconhecimento de 20 a 30 pares de bases (pb) envolvidas na duplicação de genes, deu-se em 19859, mas foi nove anos mais tarde, com a descoberta das DSB, que estas enzimas foram associadas pela primeira vez à edição genética8. Após DSB, a maquinaria de reparação do DNA da célula repara a quebra: ou por ligação de extremidade não homóloga (Non-Homologous End Joining, NHEJ), inserindo ou retirando nucleótidos, o que pode levar a um knock-out do gene onde se deu a quebra; ou por reparação direcionada por homologia (Homology-Directed Repair, HDR), inserindo um fragmento de DNA fornecido à célula. No entanto, esta técnica envolve a síntese de uma meganuclease específica de cada fragmento de DNA alvo6. Também em 1985, foram descobertas pequenas proteínas que, devido à presença de um ião de zinco que estabiliza a ligação ao DNA e de um conjunto de motivos que se ligam a sequências de três pares de bases específicas do DNA alvo, apresentam uma elevada versatilidade na ligação ao DNA10, 11. Em 1996, com a fusão destas proteínas a domínios de clivagem de DNA, foram criadas Zinc Finger Nucleases (ZFN)12. Em 2010, novas nucleases programáveis foram concebidas, as nucleases efetoras semelhantes a ativadores de transcrição (Transcription Activator-Like Effector Nucleases, TALEN) 13, enzimas que juntam, também, enzimas de clivagem de DNA a módulos de ligação a nucleótidos de DNA14, sendo que nesta técnica cada módulo liga-se a um nucleótido, usando para tal apenas dois aminoácidos, o que torna a conceção destas nucleases bastante mais fácil do que as ZFN15. Todas estas técnicas envolvem a morosa tarefa de construção de novos domínios ou módulos proteicos para cada sequência alvo a editar6. Contudo, um novo sistema viria resolver este problema e revolucionar o mundo da engenharia genética, a técnica CRISPR/Cas.
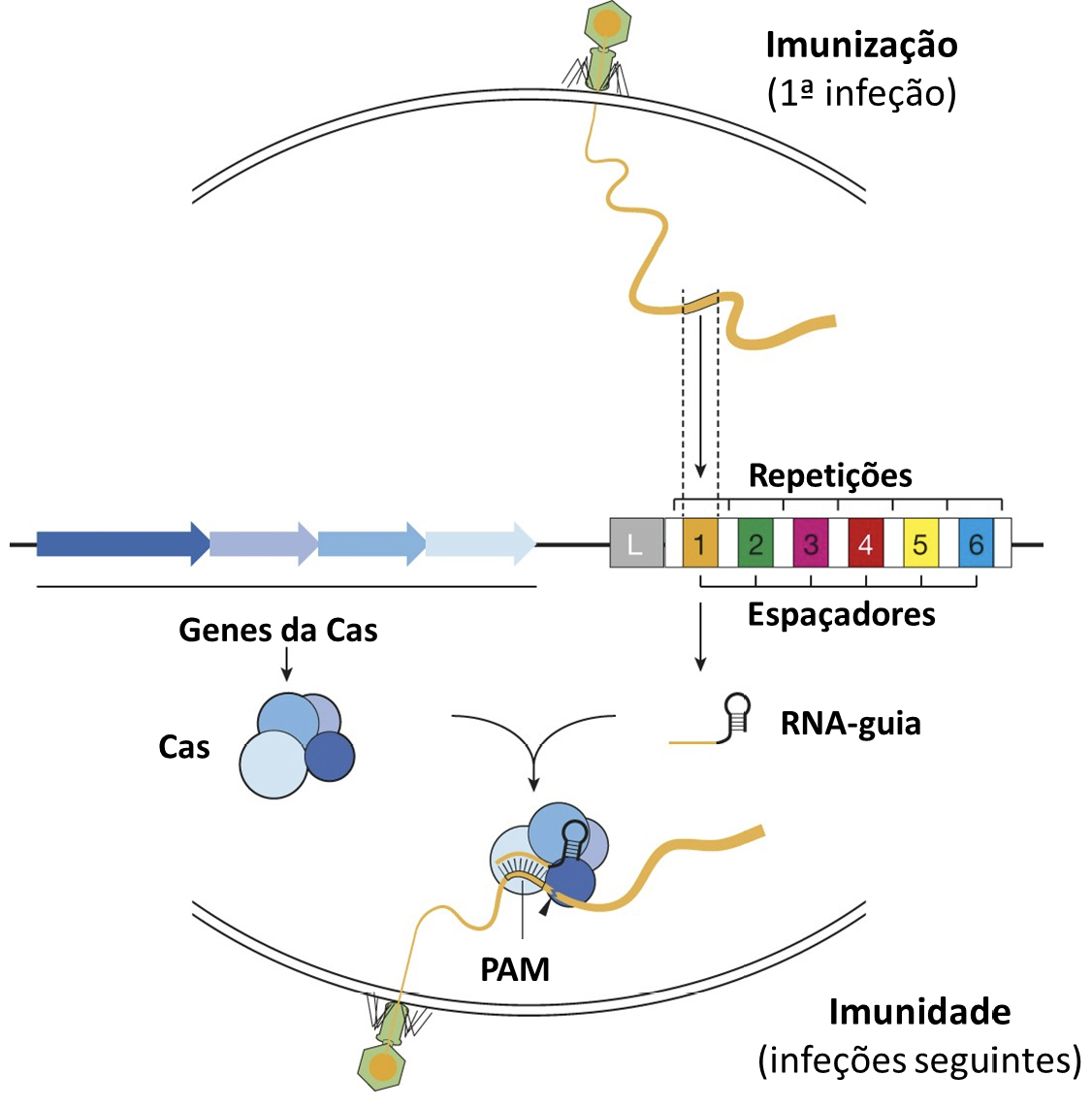
Nos finais da década de 80 do século XX, foram observadas sequências de DNA repetidas no genoma de Escherichia coli, designadas repetições, e espaçadas regularmente por sequências não repetidas derivadas de vírus, os espaçadores16. Mais tarde, aquelas repetições foram também identificadas noutros genomas de bactérias e de Archaea (arqueias)17 e associadas a genes conservados que expressam endonucleases, passando este conjunto de repetições a ser denominado de Repetições Palindrómicas Curtas Agrupadas e Regularmente Interespaçadas (Clustered Regularly Interspaced Short Palindromic Repeats, CRISPR)18. Posteriormente, os espaçadores foram, também, encontrados em genomas de fagos e noutros elementos genéticos estranhos19, 20, 21, levando a que, por fim, o conjunto de repetições CRISPR fosse associado a um sistema de defesa bacteriano contra material genético estranho. Neste sistema, numa primeira infeção por uma dado material genético estranho (fase de imunização), este é fragmentado e inserido entre as repetições, constituindo os tais espaçadores do CRISPR que, numa segunda infeção pelo mesmo material genético (fase de imunidade), irão direcionar a nuclease Cas para o genoma invasor, cortando-o de forma precisa22. Para tal, a enzima Cas usa como guia um RNA transcrito dos espaçadores inseridos no seu genoma na fase de imunização, o RNA-guia, que então se liga por complementaridade ao DNA alvo da Cas, o material genético estranho desta segunda infeção23, 24. Para que a enzima Cas o reconheça, este DNA alvo tem de estar adjacente a um motivo protoespaçador (Protospacer Adjacent Motif, PAM), fulcral para a clivagem do DNA alvo e para distinguir este DNA dos segmentos anteriormente incluídos no CRISPR25, 26, 27, 28 (FIGURA 1). Este sistema foi posteriormente adaptado para outras espécies e testado in vitro e in vivo29, 30, 31, 32, 33, 34, constituindo assim uma tecnologia de edição de genomas bastante mais prática e económica do que as anteriores, pois apenas envolve a síntese de uma pequena molécula de RNA-guia para cada alvo a ser editado, tendo sido utilizado na edição de genomas de praticamente todos os organismos modelo6.
Relativamente à aplicação da técnica CRISPR/Cas na edição de genomas de espécies vegetais, este sistema tem sido utilizado para melhorar diversas características, como a produtividade e a gestão de stresse biótico e abiótico, em plantas com interesse económico, como o arroz, a cevada, o milho, o trigo, o algodão, o tomateiro, a soja, citrinos, a batateira, a videira, a melancia, a bananeira, a cenoura, leguminosas, a mandioca e a batata-doce35. A expressão do gene da nuclease Cas, bem como dos RNA-guia nas células alvo, é suficiente para a modificação do genoma de interesse. Em 2013 iniciaram-se as primeiras edições de genomas de plantas, quer em dicotiledóneas com Arabidopsis thaliana36, uma planta modelo, quer em monocotiledóneas com o arroz, Oryza sativa37, 38. Enquanto que em Arabidopsis a primeira edição genética com esta tecnologia se centrou em provar que também funciona em plantas, tendo os autores mutado com elevada eficiência três genes relacionados com a fenologia da planta36, no caso do arroz, embora o objetivo da investigação fosse também a prova de conceito, a função dos genes mutados está associada ao controlo de respostas a stresses bióticos e abióticos37, 38.
Para além do seu interesse económico, o tomateiro é uma espécie ideal para testar técnicas de edição genómica dada a disponibilidade de métodos de transformação eficazes, a caracterização genómica já alcançada nesta espécie e a existência de um grande historial de melhoramento40. No que toca ao melhoramento de qualidade, esta planta já foi alvo de várias intervenções usando a técnica CRISPR/Cas, como obtenção de frutos partenocárpicos41, 42, aumento do tempo de prateleira43 e aumento de produtividade44, 45. Num dos estudos mais impactantes de edição do genoma do tomateiro, foi possível através da técnica CRISPR/Cas mutar a zona autoinibitória da enzima descarboxilase do glutamato46, responsável pela produção de ácido gama-aminobutírico (Gamma-AminoButyric Acid, GABA) a partir do glutamato, levando à acumulação de GABA no tomate. GABA é um aminoácido não proteico com benefícios para a tensão arterial. Um estudo posterior envolveu a criação de um mutante para quatro genes relacionados com o metabolismo do GABA, levando também à sua acumulação47. Graças a estes melhoramentos, o tomate enriquecido em GABA foi o primeiro alimento editado pela técnica CRISPR/Cas a entrar no mercado48. No controlo de respostas a stresses, a utilização de dois RNA-guia na edição do gene Mildew Resistant Locus O (MLO), gene com um grande impacto na suscetibilidade ao fungo Oidium neolycopersici, fungo responsável pela doença do oídio no tomateiro, permitiu uma deleção de 48 pb neste gene e, consequentemente, a resistência a este fungo, criando a variedade não transgénica Tomelo45, 49.
No entanto, a técnica CRISPR/Cas apresenta alguns desafios. Já na fase inicial de experiências de edição genómica por CRISPR/Cas duas abordagens foram testadas relativamente à transformação de plantas com o vetor que contém o gene da Cas e os RNA-guia: em Arabidopsis o método optado foi o floral dip50, segundo o qual os botões florais nas suas fases iniciais de desenvolvimento são mergulhados numa solução contendo bactérias da espécie Agrobacterium tumefaciens previamente transformadas com o plasmídeo contendo os genes que expressam a endonucleases Cas9 e os RNA-guia utilizados36; por outro lado, nos primeiros estudos com o arroz, o método seguido foi a transformação química de protoplastos37, 38 e o bombardeamento de calli37. Aqui encontramos um dos desafios da técnica CRISPR/Cas, a escolha do método de transformação da espécie desejada, que terá em conta quer o processo de regeneração, quer a transmissão da mutação induzida às gerações seguintes. Geralmente, as mutações originadas em Arabidopsis por esta técnica, quando o método de transformação utilizado é o floral dip, apresentam uma baixa taxa de transmissão às gerações seguintes, devido à utilização de promotores constitutivos na regulação da expressão da enzima Cas51. A adoção de enzimas Cas, cujo gene é regulado por um promotor específico das células germinativas, resolveu este problema52. Por outro lado, no caso descrito no arroz a fase de transformação ocorreu em culturas de tecidos utilizando a totipotência das células para potenciar a transmissão da modificação51. No entanto, nem todos as plantas de interesse económico são facilmente regeneradas a partir de culturas de tecidos, como as leguminosas, tendo já sido desenvolvidos diferentes protocolos otimizados de regeneração em diferentes espécies53. Para espécies recalcitrantes têm sido desenvolvidos novos sistemas vetoriais dos genes necessários ao CRISPR/Cas, como a edição genómica induzida por genes virais, onde vetores baseados em vírus são replicados na planta infetada, propagando a mutação por todo a planta incluindo nas células germinativas, e a edição genómica de linhas elite por cruzamento com linhas mutadas menos recalcitrantes54. Relativamente ao complexo Cas-RNA-guia e à sua especificidade, as mutações efetuadas pela enzima Cas em genes inespecíficos constituem um dos grandes problemas da técnica e apoiam alguns argumentos éticos contra a sua utilização, principalmente quando empregue em espécies com genomas grandes e onde a totalidade do genoma não é conhecida55. Embora os RNA-guia contenham 20 nucleótidos o mais específicos possível para o gene que se pretende mutar, outros aspetos, como a estrutura do RNA-guia, o local PAM escolhido e a estrutura final do complexo RNA-guia – Cas-gene alvo podem afetar a especificidade do corte efetuado pela endonuclease. A utilização de endonucleases modificadas de elevada fidelidade, rácios Cas/RNA-guia otimizados, plataformas de desenho de RNA-guia mais fiéis e genomas de referência de elevada qualidade podem solucionar este problema. Também a introdução de genes estranhos por HDR após o corte da Cas apresenta grandes dificuldades em eucariotas no que toca à precisão e exatidão da inserção. Algumas das soluções consistem em desligar por mutagénese os mecanismos de NHEJ, ou expressar enzimas de modificação do DNA no genoma de interesse, com o intuito de modificar a sua tipologia e facilitar a HDR, ou ainda a utilização de outros estimulantes deste processo51.
Em conclusão, embora os organismos modificados pela técnica CRISPR/Cas sejam considerados organismos geneticamente modificados (OGMs) de acordo com a legislação em vigor na União Europeia, o facto é que, ao contrário dos organismos transgénicos em que um gene com função conhecida é transferido de um organismo para o outro, sendo integrado aleatoriamente no genoma do segundo, nos organismos geneticamente editados segmentos muitos específicos do seu DNA são alterados com vista a alterar a função ou expressão do gene alvo, não havendo necessariamente inserção de um gene estranho no genoma do organismo. Aparentemente a técnica CRISPR/Cas apresenta apenas duas grandes preocupações quanto à sua aplicação em biotecnologia vegetal e no melhoramento de plantas: as mutações inespecíficas e a deriva genética. Quanto às primeiras, e como já foi referido, para além desta técnica apresentar uma maior fidelidade relativamente às técnicas de edição genómica anteriores, são atualizadas com frequência novas ferramentas que aumentam a fidelidade da técnica e novos genomas são conhecidos mais profundamente, evitando os erros das mutações inespecíficas. Quanto à deriva genética, embora a introdução de edições no genoma transmissíveis à descendência possa alterar drasticamente o pool genético e causar distúrbios em espécies silvestres, a reversão desta situação com a reintrodução da sequência original do gene editado pode resolver este problema57. Assim, embora alguns pormenores e especificidades da técnica CRISPR/Cas possam ser melhoradas, a potencialidade que apresenta para a modificação de genomas será muito importante no melhoramento de plantas num futuro próximo, principalmente com o aumento do número de espécies vegetais com o genoma sequenciado.
Referências
- 1 CAPECCHI, M. R., Altering the genome by homologous recombination, Science, 244, 1288-1292. 1989.
- 2 KHALIL, A. M., The genome editing revolution, Journal of Genetic Engineering and Biotechnology, 18, 1-16. 2020.
- 3 TUTEJA, N. et al., Recent advances in development of marker-free transgenic plants: regulation and biosafety concern, Journal of Biosciences, 37, 167-197. 2012.
- 4 CHEN, L., A method for the production and expedient screening of CRISPR/Cas9-mediated non-transgenic mutant plants, Horticulture Research, 5. 2018.
- 5 KOUL, B., Cisgenics and Transgenics: Strategies for Sustainable Crop Development and Food Security, Springer, p. 107-129. 2022.
- 6 TRÖDER, S. E. & ZEVNIK, B., History of genome editing: From meganucleases to CRISPR, Laboratory Animals, 56, 60-68. 2022.
- 7 SHUVALOV, O. et al., Current genome editing tools in gene therapy: new approaches to treat cancer, Current Gene Therapy, 15, 511-529. 2015.
- 8 ROUET, P. et al., Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease, Molecular and Cellular Biology, 14, 8096-8106. 1994.
- 9 JACQUIER, A. & DUJON, B., An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene, Cell, 41, 383-394. 1985.
- 10 KLUG, A., The discovery of zinc fingers and their applications in gene regulation and genome manipulation, Annual Review of Biochemistry, 79, 213-231. 2010.
- 11 MILLER, J. et al., Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes,, The EMBO Journal, 4, 1609-1614. 1985.
- 12 KIM, Y-G. et al., Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain, Proceedings of the National Academy of Sciences, 93, 1156-1160. 1996.
- 13 CHRISTIAN, M. et al., Targeting DNA double-strand breaks with TAL effector nucleases, Genetics, 186, 757-761. 2010.
- 14 MILLER, J. C., A TALE nuclease architecture for efficient genome editing, Nature Biotechnology, 29, 143-148. 2011.
- 15 BOGDANOVE, A. J. & VOYTAS, D. F., TAL effectors: customizable proteins for DNA targeting, Science, 333, 1843-1846. 2011.
- 16 ISHINO, Y. et al., Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product, Journal of Bacteriology, 169, 5429-5433. 1987.
- 17 MOJICA, F. J. et al., Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria, Molecular Microbiology, 36, 244-246. 2000.
- 18 JANSEN, R. et al., Identification of genes that are associated with DNA repeats in prokaryotes, Molecular Microbiology, 43, 1565-1575. 2002.
- 19 BOLOTIN, A. et al., Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin, Microbiology, 151, 2551-2561. 2005.
- 20 MOJICA, F. J. et al., Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements, Journal of Molecular Evolution, 60, 174-182. 2005.
- 21 POURCEL, C. et al., CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies, Microbiology, 151, 653-663. 2005.
- 22 BARRANGOU, R. et al., CRISPR provides acquired resistance against viruses in prokaryotes, Science, 315, 1709-1712. 2007.
- 23 BROUNS, S. J. et al., Small CRISPR RNAs guide antiviral defense in prokaryotes, Science, 321, 960-964. 2008.
- 24 MARRAFFINI, L. A. & SONTHEIMER, E. J., CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA, Science, 322, 1843-1845. 2008.
- 25 DEVEAU, H. et al., Phage response to CRISPR-encoded resistance in Streptococcus thermophilus, Journal of Bacteriology, 190, 1390- 1400. 2008.
- 26 JINEK, M. et al., A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity, Science, 337, 816-821. 2012.
- 27 MARRAFFINI, L. A. & SONTHEIMER, E. J., Self versus non-self discrimination during CRISPR RNA-directed immunity, Nature, 463, 568-571. 2010.
- 28 MOJICA, F. J. et al., Short motif sequences determine the targets of the prokaryotic CRISPR defence system, Microbiology, 155, 733-740. 2009.
- 29 CONG, L. et al., Multiplex genome engineering using CRISPR/Cas systems, Science, 339, 819-823. 2013.
- 30 GASIUNAS, G. et al., Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria, Proceedings of the National Academy of Sciences, 109, E2579-E2586. 2012.
- 31 JINEK, M. et al., RNA-programmed genome editing in human cells, Elife, 2, e00471. 2013.
- 32 MALI, P. et al., RNA-guided human genome engineering via Cas9, Science, 339, 823-826. 2013.
- 33 WANG, H. et al., One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering, Cell, 153, 910-918. 2013.
- 34 YANG, H. et al., One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering, Cell, 154, 1370-1379. 2013.
- 35 JAGANATHAN, D. et al., CRISPR for crop improvement: an update review, Frontiers in Plant Science, 9, 985. 2018.
- 36 FENG, Z. et al., Efficient genome editing in plants using a CRISPR/Cas system, Cell Research, 23, 1229-1232. 2013.
- 37 SHAN, Q. et al., Targeted genome modification of crop plants using a CRISPR-Cas system, Nature Biotechnology, 31, 686-688. 2013.
- 38 XIE, K. & YANG, Y., RNA-guided genome editing in plants using a CRISPR–Cas system, Molecular Plant, 6, 1975-1983. 2013.
- 39 MARRAFFINI, L. A., CRISPR-Cas immunity in prokaryotes, Nature, 526, 55-61. 2015.
- 40 PAN, C. et al., CRISPR/Cas9-mediated efficient and heritable targeted mutagenesis in tomato plants in the first and later generations, Scientific Reports, 6, 1-9. 2016.
- 41 KLAP, C. et al., Tomato facultative parthenocarpy results from Sl AGAMOUS-LIKE 6 loss of function, Plant Biotechnology Journal, 15, 634-647. 2017.
- 42 UETA, R. et al., Rapid breeding of parthenocarpic tomato plants using CRISPR/Cas9, Scientific Reports, 7, 1-8. 2017.
- 43 YU, Q. et al., CRISPR/Cas9-induced targeted mutagenesis and gene replacement to generate long-shelf life tomato lines, Scientific Reports, 7, 1-9. 2017.
- 44 SOYK, S., & MÜLLER, N. A. et al., Variation in the flowering gene SELF PRUNING 5G promotes day-neutrality and early yield in tomato, Nature Genetics, 49, 162-168. 2017.
- 45 PIMENTEL, D. & FORTES, A. M., Targeted genome editing using CRISPR-Cas9: applications in fruit quality and stress resilience, Advancement in crop improvement techniques, Woodhead Publishing, Elsevier, p 199-207. 2020.
- 46 NONAKA, S. et al., Efficient increase of aminobutyric acid (GABA) content in tomato fruits by targeted mutagenesis, Scientific Reports, 7, 1-14. 2017.
- 47 LI, R. et al., Multiplexed CRISPR/Cas9-mediated metabolic engineering of aminobutyric acid levels in Solanum lycopersicum, Plant Biotechnology Journal, 16, 415-427. 2018.
- 48 WALTZ, E., GABA-enriched tomato is first CRISPR-edited food to enter market, Nat Biotechnol, 40, 9-11. 2022.
- 49 NEKRASOV, V. et al., Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion, Scientific Reports, 7, 1-6. 2017.
- 50 CLOUGH, S. J. & BENT, A. F., Floral dip: a simplified method for Agrobacterium mediated transformation of Arabidopsis thaliana, The Plant Journal, 16, 735-743. 1998.
- 51 PAUL, J. W. & QI, Y., CRISPR/Cas9 for plant genome editing: accomplishments, problems and prospects, Plant Cell Reports, 35, 1417- 1427. 2016.
- 52 WANG, Z-P. et al., Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation, Genome Biology, 16, 1-12. 2015.
- 53 BHOWMIK, P. et al., CRISPR/Cas9 gene editing in legume crops: opportunities and challenges, Legume Science, 3, e96. 2021.
- 54 SON, S. & PARK, S. R., Challenges facing CRISPR/Cas9-based genome editing in plants, Frontiers in Plant Science, 13, 902413. 2022.
- 55 ZHANG, F. et al., CRISPR/Cas9 for genome editing: progress, implications and challenges, Human Molecular Genetics, 23, R40-R46. 2014.
- 56 MANGHWAR, H. et al., CRISPR/Cas systems in genome editing: methodologies and tools for sgRNA design, off-target evaluation, and strategies to mitigate off-target effects, Advanced Science, 7, 1902312. 2020.
- 57 ES, I., The application of the CRISPR-Cas9 genome editing machinery in food and agricultural science: current status, future perspectives, and associated challenges, Biotechnology Advances, 37, 410-421. 2019.
Este artigo já foi visualizado 8281 vezes.