

Espectroscopia de Raman
📧 , 📧 , 📧 , 📧
- * U. Aveiro
- ɫ U. Aveiro
- ‡ U. Aveiro
- + U. Aveiro
Referência Claro, P. R., Nolasco, M. M., Araújo, C., Martins, J. T. S., (2026) Espectroscopia de Raman, Rev. Ciência Elem., V14(1):002
DOI http://doi.org/10.24927/rce2026.002
Palavras-chave
Resumo
A espectroscopia de difusão de Raman, ou apenas "espectroscopia de Raman", é uma técnica baseada no espalhamento ou difusão inelástica da luz (efeito Raman, assim designado em homenagem a C. V. Raman, 1888–1970). Embora possa ser usada para observar movimentos de baixa frequência de um sistema (incluindo movimentos rotacionais e libracionais), é normalmente descrita como uma técnica de espectroscopia vibracional1, 2.
Enquanto técnica de espectroscopia vibracional, a espectroscopia de Raman fornece informação sobre os níveis de energia dos modos normais de vibração de um sistema – os mesmos modos vibracionais estudados pela espectroscopia de absorção no infravermelho. Como os níveis vibracionais dependem da composição do sistema (tipo de átomos, força das ligações, geometria, interações intermoleculares, etc.), tanto os espectros de Raman como os de infravermelho funcionam como uma “impressão digital” da estrutura química, permitindo identificar e caracterizar uma amostra.
A diferença entre a espectroscopia de difusão de Raman e a espectroscopia de absorção no infravermelho resulta das diferenças nos processos físicos que governam a interação entre a radiação e a matéria. Tal como ilustrado na FIGURA 1 A), a espectroscopia no infravermelho mede diretamente a diferença de energia entre níveis vibracionais: para que ocorra absorção, a energia da radiação incidente tem de coincidir exatamente com a diferença de energia entre o nível vibracional fundamental e o nível excitado (identificados como v=0 e v=1 na FIGURA 1).
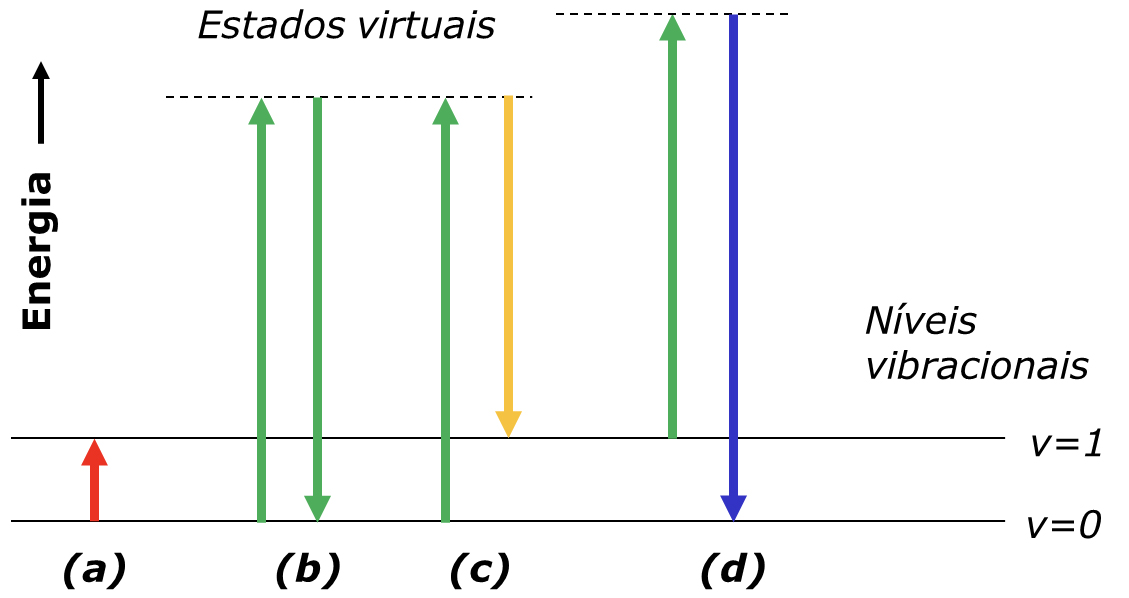
Para provocar a difusão de Raman, a amostra é iluminada com radiação monocromática cuja energia é superior à diferença entre v=0 e v=1. A descrição física do processo assume que o fotão incidente é absorvido, levando o sistema a um estado de energia virtual (linhas tracejadas na FIGURA 1), a partir do qual o fotão difundido é imediatamente emitido. Os fotões incidentes podem ser difundidos de forma elástica (difusão de Rayleigh, FIGURA 1 B)) ou inelástica (difusão de Raman, FIGURAS 1 C) e 1 D)). A diferença de energia entre os fotões incidentes e os fotões difundidos de forma inelástica, \( \nu -\nu_0\), chama-se desvio de Raman e corresponde à mesma diferença de energia medida diretamente na absorção no infravermelho.
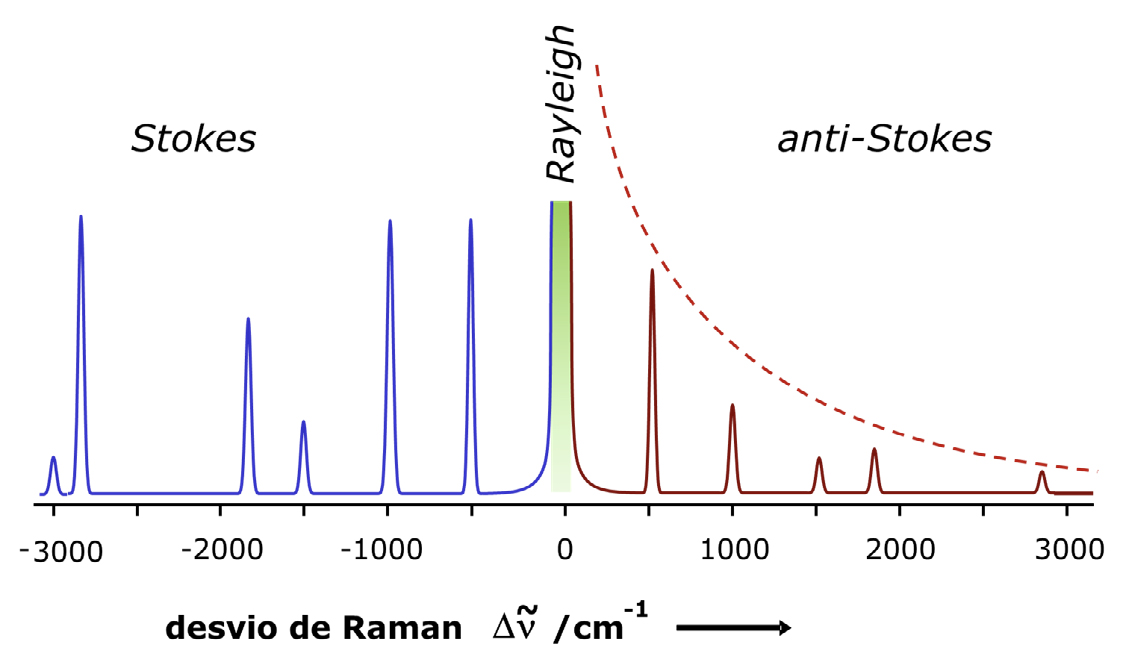
Quando os fotões difundidos têm menor energia do que os incidentes, os desvios observados são negativos e correspondem aos desvios de Stokes, ou região de Stokes (FIGURA 1 C)). Os desvios de anti-Stokes (FIGURA 1 D)) correspondem a valores positivos de ν − ν0, uma vez que o nível inicial é v=1 ou superior. Normalmente, o espectro de Raman é registado apenas na região de Stokes, pois a região anti-Stokes contém a mesma informação sobre os níveis de energia, mas apresenta menor sensibilidade: a intensidade das transições anti-Stokes diminui exponencialmente com o aumento de \( \nu -\nu_0\), devido à menor população dos estados vibracionais excitados v=1 (FIGURA 2).
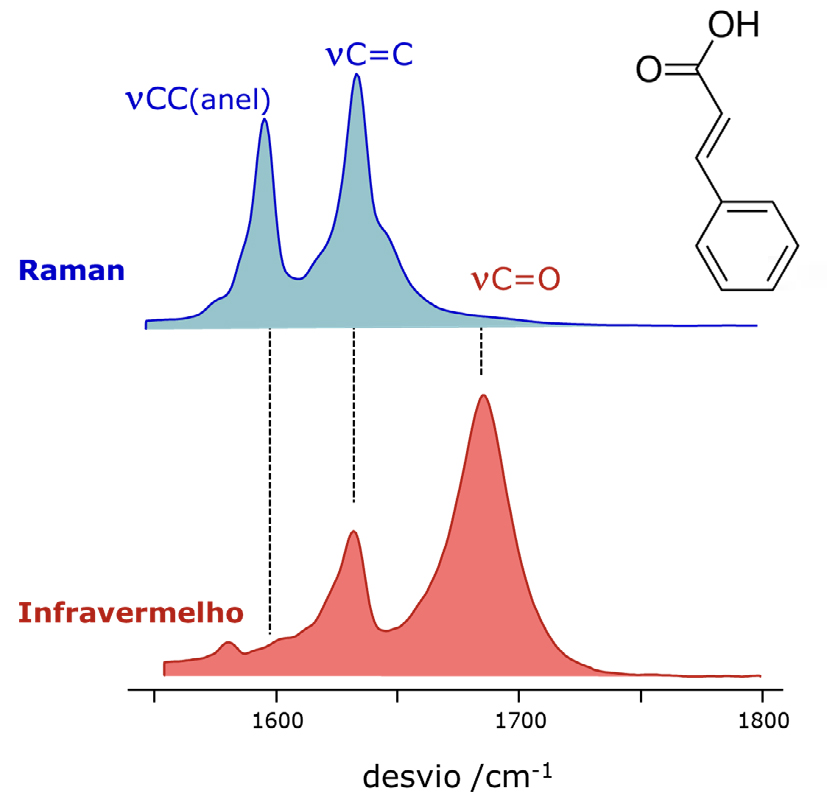
Na espectroscopia de absorção no infravermelho, a intensidade das bandas é determinada pela variação do momento dipolar durante o movimento vibracional. Uma grande variação do momento dipolar resulta numa absorção intensa; se não houver variação do momento dipolar durante a vibração, não ocorre absorção. Isto explica porque o modo de estiramento do grupo carbonilo, \(\nu\)(C=O), origina a conhecida banda intensa por volta de 1750 ± 90 cm-1, característica de compostos orgânicos como aldeídos, cetonas e ácidos carboxílicos, enquanto o modo de estiramento C=C é geralmente descrito como uma banda de absorção fraca.
Na espectroscopia de Raman, a intensidade da interação entre a radiação e a matéria – isto é, o número de fotões difundidos por um determinado modo vibracional – depende da variação da polarizabilidade molecular durante o movimento vibracional. A polarizabilidade, que traduz a facilidade de distorção da nuvem electrónica, tem de variar de forma assimétrica ao longo da vibração. Este conceito é um pouco menos intuitivo do que a “variação do momento dipolar”, mas o resultado prático é que os modos fortemente ativos no infravermelho tendem a ser fracos em Raman, enquanto os modos fracos ou mesmo invisíveis no infravermelho aparecem frequentemente como bandas intensas no espectro de Raman.
A FIGURA 3 ilustra este comportamento ao comparar os espectros de infravermelho e de Raman do ácido trans-cinâmico, um composto orgânico com ligações C=O e C=C duplas: no espectro de Raman, a banda mais intensa corresponde ao modo \(\nu\)(C=C), enquanto a banda \(\nu\)(C=O), dominante no espectro de infravermelho, é praticamente inexistente.
Isto significa que, para estudos estruturais, estas duas técnicas são complementares – e são frequentemente usadas em conjunto. No entanto, do ponto de vista analítico, é importante ter presente que os padrões de bandas que caracterizam um determinado sistema são diferentes em absorção no infravermelho e em difusão de Raman. Para um espectroscopista vibracional, a escolha entre estas técnicas deve basear-se nas suas vantagens relativas face ao objetivo pretendido.
De acordo com a descrição anterior, a espectroscopia de Raman requer uma fonte de radiação monocromática – a frequência \(\nu_0\) em relação à qual são medidos os desvios Raman. Como o efeito Raman é muito fraco, é desejável também um feixe intenso, idealmente com trajetória pouco divergente. As características “monocromático”, “não divergente” e “alta intensidade” descrevem quase perfeitamente a radiação LASER (o LASER acrescenta ainda a coerência do feixe – todas as ondas elementares emitidas mantêm uma diferença de fase constante no espaço e no tempo. Por esta razão, os espectrómetros de Raman utilizam fontes laser.
Vale a pena introduzir aqui uma nota histórica: C. V. Raman observou pela primeira vez o “efeito Raman” usando um feixe de luz solar filtrado e colimado (com seleção de raios de trajetória paralela). O equipamento incluía um telescópio para recolher a luz difundida pela amostra, filtros de vidro colorido para selecionar o comprimento de onda da luz incidente e bloquear a difusão de Rayleigh, e os próprios olhos de Raman como detetor. O primeiro “espectro de Raman” foi obtido por C. V. Raman com uma lâmpada de arco de mercúrio em quartzo, um filtro azul para selecionar a linha de 435,8 nm e uma placa fotográfica como sistema de registo.
A frequência da fonte laser, \(\nu_0\), tem várias implicações práticas (por exemplo, tamanho do foco do feixe, profundidade de penetração na amostra, intensidade da luz difundida, entre outras), mas há uma que merece destaque: quando \(\nu_0\) é elevada, o estado virtual pode coincidir com um estado electrónico excitado da amostra, originando fluorescência (FIGURA 4 C)). O efeito Raman de ressonância, também representado, é um caso particular do efeito Raman que produz uma amplificação muito forte da difusão associado aos modos vibracionais ligados à alteração geométrica da molécula no estado electrónico excitado.
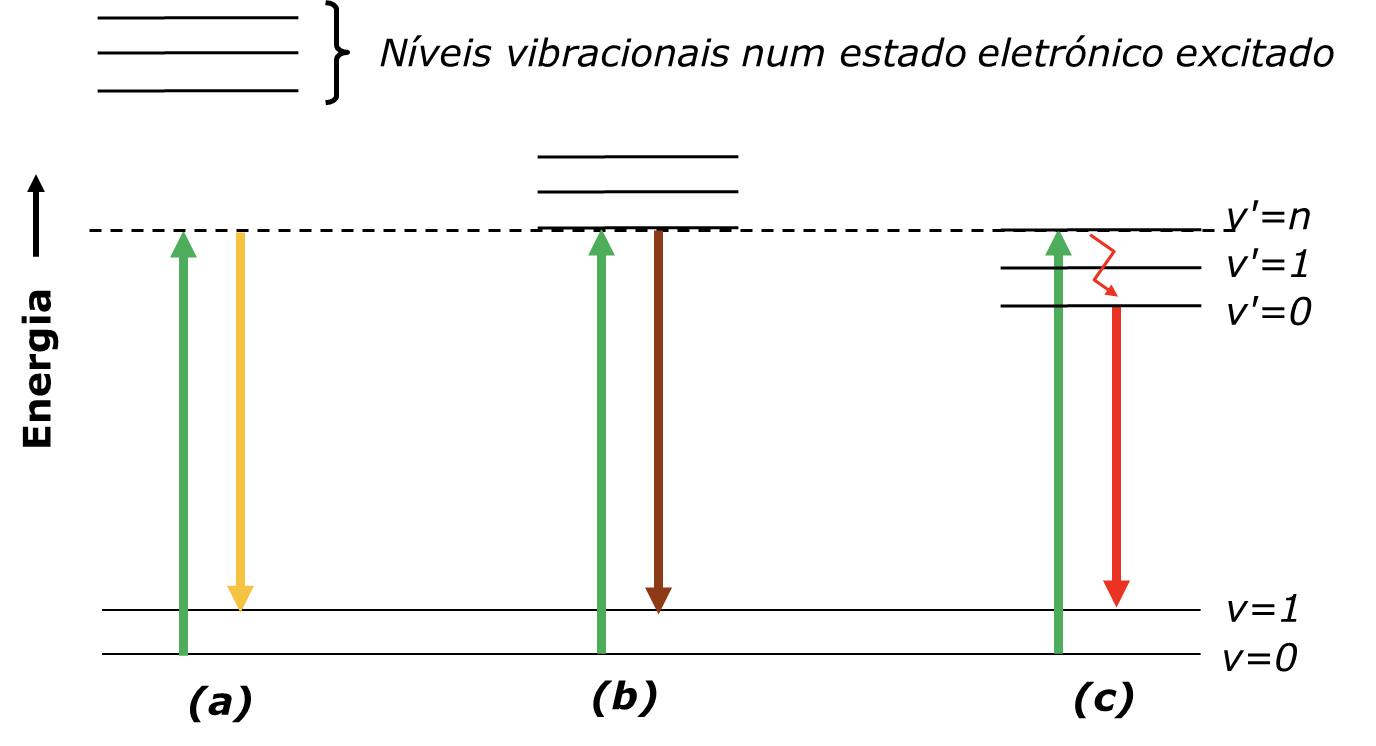
A fluorescência é várias ordens de grandeza mais intensa do que o efeito Raman, o que constitui um problema mesmo quando — e muitas vezes precisamente quando — o sistema fluorescente é apenas uma impureza da amostra. Até há poucas décadas, eram necessários processos de purificação longos e trabalhosos para eliminar a fluorescência causada por impurezas. Este foi o principal motivo que impediu a espectroscopia de Raman de competir com a espectroscopia no infravermelho como técnica de rotina em muitas aplicações práticas (por exemplo, química alimentar, indústria petroquímica, indústria da pasta e papel, aplicações biomédicas, entre outras).
A solução para este problema passa por afastar a energia do laser das transições electrónicas indesejadas. Isto é normalmente conseguido alterando a frequência da fonte laser (por exemplo, da linha verde de comprimento de onda 514,5 nm de um laser Ar+ para a região do infravermelho próximo, de um laser Nd:YAG com radiação de comprimento de onda 1064 nm). A desvantagem dos lasers de maior comprimento de onda é a sensibilidade do detetor, que geralmente diminui com o aumento do comprimento de onda (ou diminuição da frequência e, portanto, da energia). Além disso, ocorre uma perda direta de sinal, uma vez que a eficiência da difusão Raman é proporcional a \(\nu_0^{4}\), sendo \(\nu_0\) a frequência da radiação incidente. Esta limitação foi ultrapassada com a aplicação da Transformada de Fourier à espectroscopia de Raman (FT-Raman). No FT-Raman, todas as frequências chegam ao detetor em simultâneo, compensando a menor sensibilidade do detetor (vantagem multiplex, ou vantagem de Fellgett).
Como resultado, lasers de frequência mais elevada são usados em espectrómetros dispersivos “convencionais” – com monocromador e detetores fotomultiplicadores ou dispositivos CCD – enquanto lasers de menor frequência são usados nos espectrómetros FT, equipados com interferómetro, filtro de rejeição de faixa (“notch”) e detetores de estado sólido.
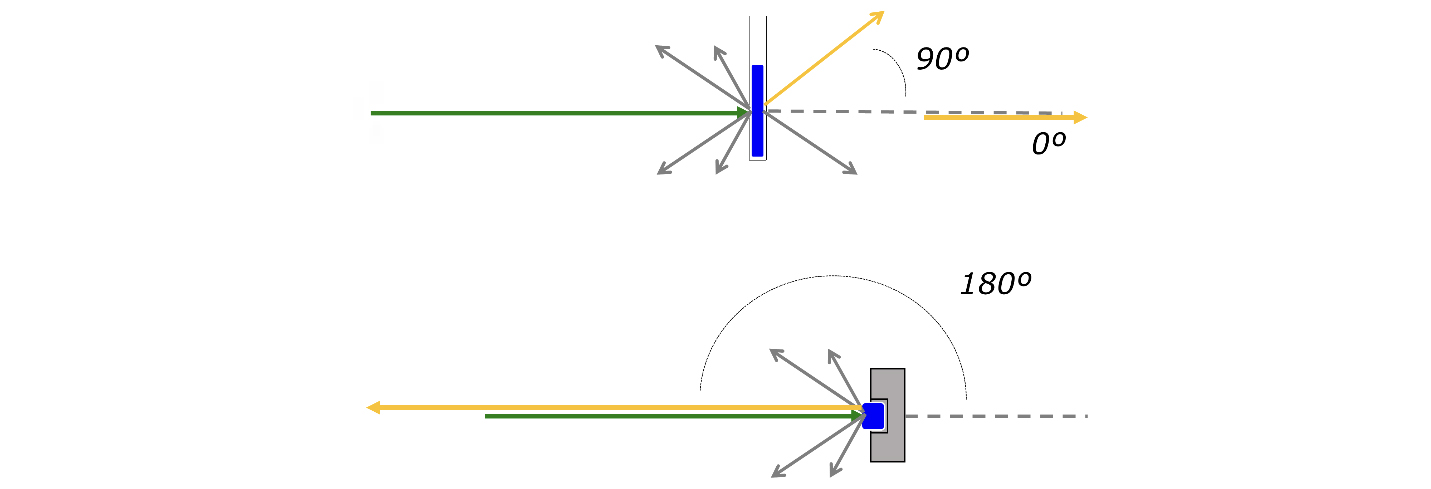
Uma característica muito importante da espectroscopia de Raman é o tamanho da amostra – ou, mais precisamente, o tamanho da porção da amostra que é efetivamente analisada. Isto tem implicações tanto no tamanho das partículas que podem ser estudadas por Raman como nas aplicações em imagem. A colocação da amostra é relativamente simples e são possíveis várias geometrias de recolha da radiação (FIGURA 5).
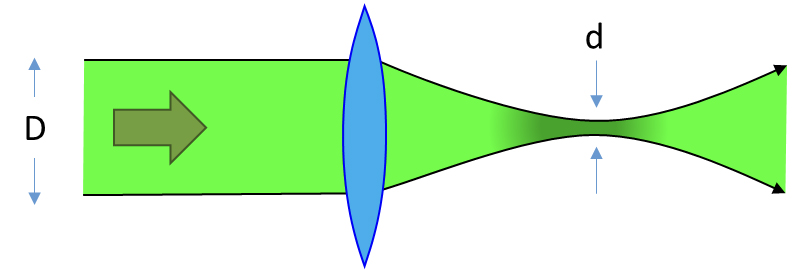
O feixe laser incidente é geralmente estreito (cerca de 1 mm) e, como ilustrado na Figura 6, um feixe com 1 mm de largura pode ser focado num ponto com diâmetro de 10 μm ou menos, dependendo do comprimento de onda. A penetração do laser na amostra depende (i) do comprimento de onda (quanto maior o comprimento de onda, maior a profundidade de penetração) e (ii) da densidade óptica da amostra. Definir um valor típico de profundidade de penetração é algo especulativo, mas um intervalo de 1 a 200 μm pode ser considerado indicativo.
Isto define um volume focal do laser, que constitui uma estimativa mínima do volume analisado. A radiação laser também é difundida para fora do volume focal, originando difusão Raman nessas regiões, pelo que o volume efetivamente analisado é maior (FIGURA 7).
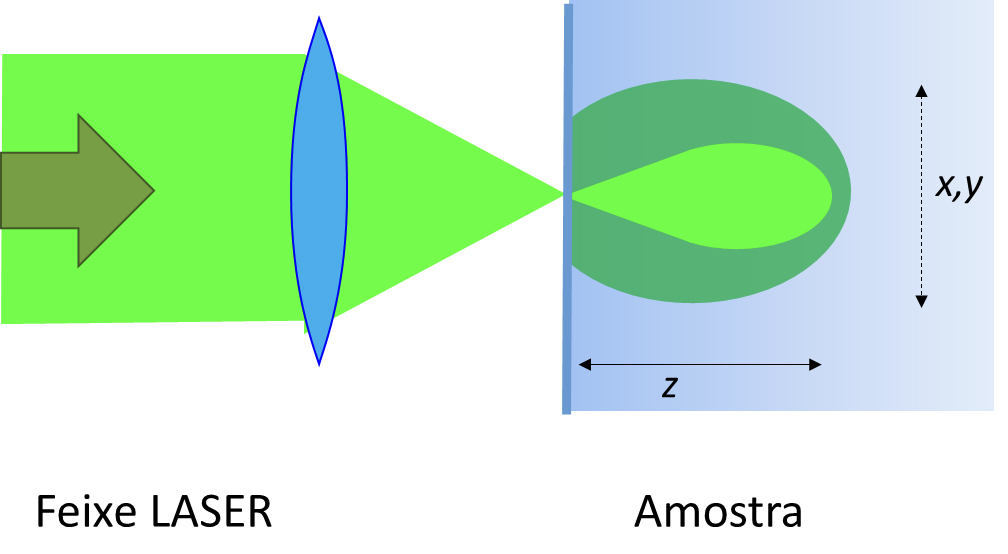
A extensão do volume analisado depende naturalmente das propriedades da amostra, mas também da óptica de recolha: uma combinação adequada de lentes de recolha e diafragmas com fenda de corte de radiação pode limitar a origem do sinal Raman que chega ao detetor, melhorando significativamente a resolução espacial do espectrómetro.
Estas possibilidades são amplamente exploradas na microscopia Raman e, de forma ainda mais eficaz, na microscopia Raman confocal. A utilização de um microscópio para focar o feixe na amostra e recolher o sinal Raman apresenta uma dupla vantagem: devido à grande abertura numérica (NA) das objectivas, o feixe laser é focado num ponto muito pequeno e a radiação difundida é recolhida num ângulo amplo (FIGURA 8). O aumento da irradiância local e a recolha em grande ângulo compensam a redução do volume de amostra analisado.
Num sistema confocal, a radiação recolhida percorre um trajeto através de diafragmas com planos opticamente conjugados, o que proporciona uma filtragem espacial muito eficiente. A radiação difundida proveniente de zonas fora de foco é rejeitada, e apenas a radiação originada num pequeno volume da amostra, dentro do volume focal, chega ao detetor.
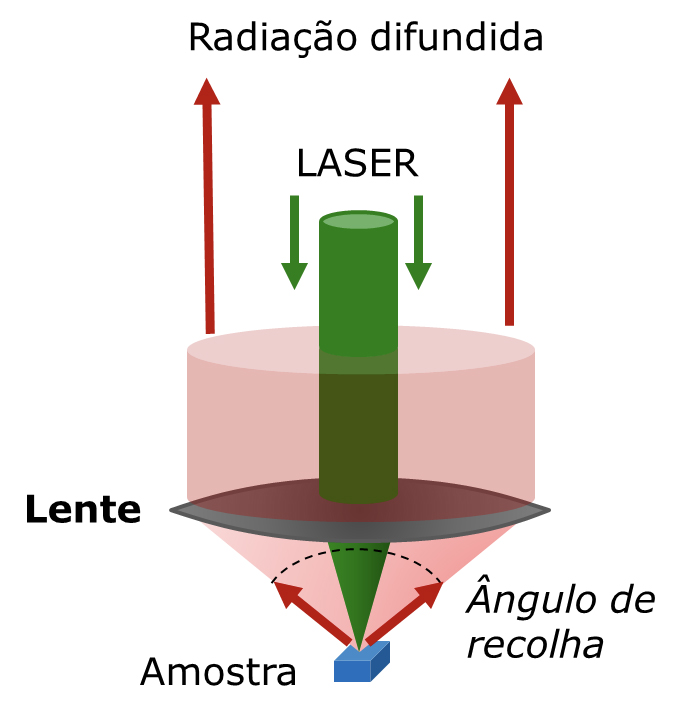
Desta forma, a microscopia Raman oferece uma resolução espacial típica de cerca de 1 μm à superfície (resolução em x-y), e a microscopia confocal acrescenta resolução em profundidade (resolução em z) de magnitude semelhante.
Referências
- 1 CLARO, P. R., Espectroscopia Vibracional, Rev. Ciência Elem., V6(2):040. 2018.
- 2 CLARO, P. R., Espectro, Rev. Ciência Elem., V6(3):055. 2018.
Este artigo já foi visualizado 794 vezes.